This is an automatically translated article.
The article was professionally consulted by resident Doctor Nguyen Quynh Giang - Department of Diagnostic Imaging and Nuclear Medicine - Vinmec Times City International Hospital. Dr. Giang has many years of experience in the field of diagnostic imaging, especially in the field of multi-segment computed tomography, magnetic resonance.Cystic fibrosis can cause infections, poor absorption of nutrients, and it can also affect the sweat glands along with the male reproductive system. To make an accurate diagnosis in addition to blood tests and sweat tests, your doctor may order an evaluation of your condition with imaging tests such as: chest X-ray, CT scan of the chest or abdomen, chest MRI, or abdomen, abdominal ultrasound or CT sinus.
1. Cystic Fibrosis
Cystic fibrosis is a life-threatening genetic disease that causes the body to produce thick mucus. This thick mucus can build up and clog ducts and other ducts in the lungs, digestive tract, and pancreas. What's more, the buildup can also cause serious infections, digestive problems, and even death. In addition, it can affect the sweat glands and male reproductive system.In cystic fibrosis, a defect (mutation) in a gene - the cystic fibrosis transmembrane transduction regulator (CFTR) gene - changes the protein that regulates salt movement in and out of cells. The result is thick, sticky mucus in the respiratory, digestive and reproductive systems, as well as increased salts in sweat. Many different defects can occur in the gene. These gene mutations are associated with disease severity.
Children must inherit a pair of gene copies from each parent in order to develop the disease. If children inherit only one copy, they will not develop cystic fibrosis. However, they will be carriers of the disease and can pass this gene on to their children.
Although cystic fibrosis can occur in people of any race, it is most common in white people of Northern or Central European descent.
Xơ nang có thể gây ra tình trạng nhiễm trùng phổi
Short of breath Frequent lung infections Persistent cough Wheezing Fatigue Nasal congestion Abdominal pain Abdominal pain Abnormal and smelly stools foul smell Constipation Bowel obstruction Male infertility There is now a need to be screened for cystic fibrosis and other conditions at birth. Therefore, infants are often diagnosed early after birth, before symptoms occur. However, it is important to recognize early symptoms in infants, including:
Salty skin Underweight Slow growth Rectal fistula.
2. Diagnosis of cystic fibrosis
The most common types of cystic fibrosis tests include taking a blood sample for genetic testing or conducting a sweat test. The sweat test measures the amount of salt in a person's sweat. The presence of high salt may indicate cystic fibrosis.Sweat chloride test The sweat chloride test is the most commonly used test to diagnose cystic fibrosis. It checks for increased salt levels in sweat. The test is performed using a chemical that causes the skin to sweat when activated by a weak electrical current. Sweat is collected on a pad or paper and then analyzed. A diagnosis of cystic fibrosis is made if the sweat is saltier than normal.
Sputum test During a sputum test, the doctor takes a sample of mucus. The sample can confirm the presence of a lung infection. In addition, it can also show the types of germs present and determine which antibiotics work best to treat them.
Immuno Trypsinogen Test (IRT) An immune trypsinogen test (IRT) is a test that checks for abnormal levels of a protein called IRT in the blood. A high level of IRT can be a sign of cystic fibrosis. However, further testing is needed to confirm the diagnosis.
Currently, to screen newborns for cystic fibrosis, an immunological trypsinogen test is required. In some cases, pregnant women can get a prenatal diagnosis through amniocentesis or a biopsy of the placenta, known as chorionic villus sampling (CVS). Amniocentesis is the removal of fluid from the amniotic sac (the fluid around the developing embryo) through a specialized needle through the abdomen. This fluid can then be tested to evaluate for cystic fibrosis. During chorionic villus sampling, a needle is used to remove a small amount of the placenta, which will then be used to evaluate for cystic fibrosis and other genetic diseases.

Một trong những xét nghiệm chẩn đoán bệnh xơ nang là xét nghiệm mồ hôi
Computed tomography of the chest or abdomen (CT) : These tests use special X-ray equipment and computers to create many detailed images. secretions in the lungs or intestines. CT scan images can help determine the severity of cystic fibrosis by looking at the amount of mucus as well as looking for dilated airways in the lungs. Furthermore, the tests can also look for infections. This computed tomography test uses ionizing radiation. Chest X-ray: This method uses a small amount of ionizing radiation to create images that help evaluate dilated airways that contain mucus and also to evaluate for lung infections that need to be treated with antibiotics. . A chest x-ray is used as a routine checkup to observe changes in patients with cystic fibrosis and to rule out other respiratory conditions such as pneumonia or a collapsed lung. Chest or abdominal magnetic resonance imaging (MRI): These imaging tests use a powerful magnet, radio waves, and a computer to create detailed images of the lungs and digestive tract. Although a chest X-ray or CT is more commonly used for this disease, an MRI can help assess the severity of cystic fibrosis. Abdominal ultrasound: This imaging test uses a small transducer, gel, and high-frequency sound waves, to create images of the upper abdomen. It helps evaluate the pancreas, liver, and gallbladder, all of which are affected by cystic fibrosis. CT of the sinuses: this test combines special X-ray equipment and a computer to create multiple images of the paranasal sinus cavities. It can help identify single nasal polyps, which are common in patients with cystic fibrosis.
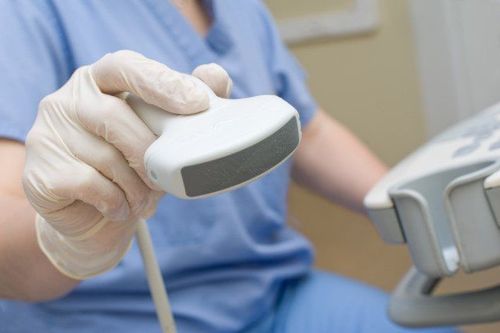
Siêu âm ổ bụng cho phép đánh giá tình trạng của bệnh xơ nang
3. Treatment of cystic fibrosis
Although there is no cure for cystic fibrosis, your doctor may prescribe one or more of the following to help relieve symptoms and improve quality of life:Lifestyle changes such as dietary supplements Better yet, take vitamin supplements, increase physical activity, avoid tobacco use, and avoid secondhand smoke. Using medications such as antibiotics and inhalers can help open the airways and clear mucus and infection. Various chest treatments or airway clearance techniques (ACT) can help loosen mucus, making it easier to cough up phlegm and clear it out of the lungs. In severe cases, surgery or other procedures may be needed to improve quality of life. These surgeries may include a lung transplant, procedures to stop bleeding from the lung, removal of nasal polyps...

Sử dụng thuốc kháng sinh đang đem lại hiệu quả cao trong điều trị bệnh xơ nang
Results of the patient's examination will be returned to the home. After receiving the results of the general health examination, if you detect diseases that require intensive examination and treatment, you can use services from other specialties at the Hospital with quality treatment and services. outstanding customer service.
Please dial HOTLINE for more information or register for an appointment HERE. Download MyVinmec app to make appointments faster and to manage your bookings easily.
Reference source: radiologyinfo.org; mayoclinic.org; healthline.com