This is an automatically translated article.
The article is professionally consulted by resident doctor Duong Van Sy - Department of Pediatrics - Neonatology - Vinmec Hai Phong International General Hospital.
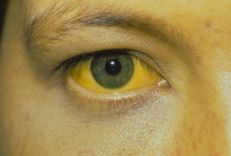
Congenital biliary atresia is a rare disease of the liver and biliary tract, characterized by disruption or deficiency of the extrahepatic biliary system, causing obstruction of bile flow. Due to misdiagnosis and late detection, many infants are missed the golden opportunity to have surgery to treat the disease. So when is the best time to treat congenital biliary atresia?
1. Treatment of congenital biliary atresia
Currently, surgery is the main treatment for congenital biliary atresia. When the disease is detected, often newborns are treated with Kasai surgery.
This is surgery to create a passage for bile from the liver using part of the small intestine. The surgery replaces the blocked bile ducts outside the liver with an appendix that acts as a new tube. The success of this surgery depends on the age of the patient (preferably 2-3 months old), the degree of liver damage and the level and experience of the surgeon.
According to the literature, this measure can reduce the symptoms of the disease, especially jaundice, for more than 60% of pediatric patients. However, some neonates after surgery have had recurrence of symptoms and severe complications due to biliary obstruction such as hepatomegaly, splenomegaly, varicose veins in the visceral organs, cirrhosis, and liver failure. , severe infections such as pneumonia, pancreatitis...
The purpose of the Kasai procedure is to allow the drainage of bile from the liver into the intestines to be guided through a new tube. If this surgical approach is performed early (before 3 months of age), the success rate is up to 80%. In infants who respond well, jaundice and other symptoms usually disappear after a few weeks. Besides, Kasai surgery is most successful in babies under 3 months of age, so early diagnosis is extremely important to save the life of the newborn.
If the Kasai procedure is not successful, the only way to treat congenital biliary atresia is a liver transplant. Liver transplantation is indicated in the treatment of congenital biliary atresia in neonates with a 10-year survival rate of more than 90%. The big problem today is the lack of grafts, especially for babies who are too young. Therefore, liver transplantation in the family, or to put it simply, a liver transplant for a child by taking half of the father's or mother's liver to transplant to the child becomes suitable in the current situation of scarce grafts. In addition, doctors and families must quickly find a suitable donor organ, before damage to the liver leads to death.
2. Best time to treat biliary atresia
Theoretically, the doctor can apply surgical treatment as soon as possible as soon as the child is found to have congenital biliary atresia. However, surgery too soon, the child will have to face the risk of rupture of the anastomosis, that is, the two mouths connecting the liver hilum and the retina are broken out, bleeding, and more complications. Therefore, experts recommend surgery for children from 1 to 2 months old. Until 100 days old, the baby still has a good chance of doing surgery. From 100 days old onwards, the more bile stasis, the more cirrhosis of the liver, so it is not good for children.

Thời điểm tốt nhất để điều trị teo đường mật ở trẻ
3. Note when treating congenital biliary atresia
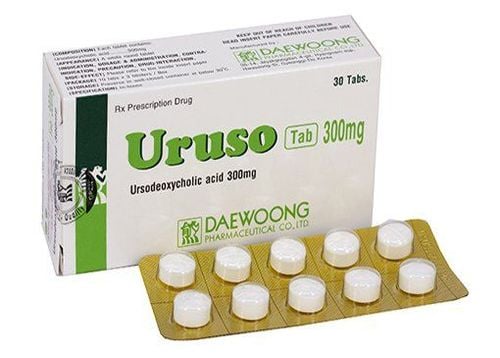
Fasting and parenteral nutrition until the child has yellow or green stools after surgery. Depending on the nature and color of stools to adjust the daily diet; Use antibiotics to prevent biliary tract infections soon after surgery, in the first 6 months after surgery with prophylactic dose Cotrimoxazol; Ursodeoxycholic (UDCA): Long-term use for 18-24 months or until the child stops cholestasis with a dose of 15-30 mg/kg/24 hours; Supplement with fat-soluble vitamins A, D, E, K daily; Use hydrolyzed protein milk with short and medium chain fatty acids; Congenital biliary atresia is a disease requiring surgery because it is the first "salvation" for newborns. Parents should note if the baby has jaundice, has a band of cement-like discolored stools from week 2 to 4 after birth, and even a few appear discolored stools right in the first few days when the meconium is gone, please take the child to the doctor early.
Please dial HOTLINE for more information or register for an appointment HERE. Download MyVinmec app to make appointments faster and to manage your bookings easily.