This is an automatically translated article.
Article by Master, Doctor Vu Duy Dung - Department of General Internal Medicine - Vinmec Times City International Hospital
Neurofibromatosis type 2 (NF2) is an autosomal dominant multisystem disease that often leads to the occurrence of multiple benign neuroendocrine tumors, central nervous system tumors, and CNS tumors. , eye abnormalities, and nerve-skin lesions.
The NF2 gene is located on chromosome 22q12 and encodes the protein merlin (also known as schwannomin), a tumor suppressor. Merlin has multiple functions involving the interactions between cell membranes and cytoskeleton proteins but, like neurofibromin in neurofibromatosis type 1 (NF1), mainly downregulates cell signaling via the RAS pathway - MAPK. Epidemiological studies indicate an incidence of NF2 of approximately 1 in 33,000 to 1 in 40,000 live births.
1. Diagnosis of neurofibromatosis type 2
Diagnostic criteria for NF2 have been improved over time to increase sensitivity as the genetic basis and important clinical features have been elucidated. The Manchester criterion is still commonly used, but in 2011 Baser and colleagues provided the current understanding to develop a criterion that could be used to diagnose earlier than the previous criterion. While vestibular schwannomas are at the core of NF2, other clinical features are often needed to confirm the diagnosis.
NF2 is most commonly seen in the second or third decade of life with hearing loss, tinnitus, or loss of balance, attributed to vestibular schwannoma. Children with NF2 are more likely to have a reason to visit for non-vestibular causes, such as brain tumors, spinal cord tumors, skin lesions, visual disturbances, and mononeuropathy .
NF2 is now rarely confused with NF1, but schwannomatosis should be included in the differential diagnosis of any patient with suspected NF2. Schwannomatosis is a rare third form of neurofibromatosis, with clinical, genetic, and imaging overlap with NF2. Mutations in the SMARCB1 gene cause 40% to 50% of familial schwannomatosis and 10% of sporadic cases. More recently, accompanying mutations in the LZTR1 gene, located on chromosome 22 in the region of the NF2 and SMARCB1 genes, have also been recognized in some cases. People with schwannomatosis typically present between the ages of 20 and 30 with chronic pain and symptoms associated with nerve sheath tumors. While other cranial nerves may be involved, individuals with schwannomatosis can be distinguished from NF2 patients because they do not have vestibular schwannoma.

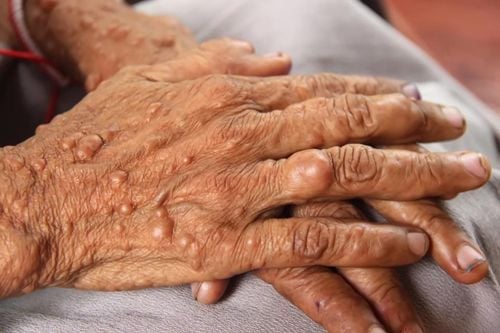
Bệnh u xơ thần kinh týp 2 là một bệnh lý đa hệ thống di truyền gen trội nhiễm sắc thể
Manchester criteria for diagnosing neurofibromatosis type 2
Patients with the following criteria may be diagnosed with neurofibromatosis type 2:
Bilateral vestibular schwannomas First-generation relatives have neurofibromatosis type 2 AND: Unilateral vestibular schwannoma OR Two of the following: meningioma, glioma, neurofibromatosis, schwannoma, posterior subcapsular cataract Vestibular schwannoma unilateral AND two of the following: meningioma, glioma, neurofibromatosis, schwannoma, posterior subcapsular cataract Multiple meningioma (≥ 2) AND: Unilateral vestibular schwannoma OR Two of the following: meningioma, glioma, neurofibromatosis, schwannoma, posterior subcapsular cataract
2. Clinical features of neurofibromatosis type 2
Clinical manifestations of NF2 involve the skin, central nervous system, peripheral nervous system, and eyes.
2.1. Skin The cutaneous symptoms of NF2 are less prominent than those of other neuro-dermatopathies. Skin tumors are found in about 70% of patients. Areas of hyperpigmentation, located in the skin, raised in patches sometimes accompanied by alopecia are most common. Palpable nodular lesions under the skin are also common. Coffee-milk spots are seen in about 40% of patients but are atypical and less numerous than with NF1.
2.2. Central and Peripheral Nervous System Vestibular schwannomas are benign tumors of the VIIIth cranial nerve and are the hallmark of NF2. They originate in the inner ear canal and extend to the pontine angle of the cerebellum. They may initially develop and become symptomatic on one side but often become bilateral over time. Vestibular schwannomas are the major cause of disability in NF2, causing neurosensory hearing loss, tinnitus, loss of balance, and brainstem compression that increases over time. Several studies of the natural history of vestibular schwannomas have shown a growth rate of 1 mm/year to 1.8 mm/year. Interestingly, the degree of hearing loss does not necessarily correlate with tumor size or tumor growth rate.
Other benign intracranial tumors are also common in NF2. After vestibular schwannomas, the trigeminal and oculomotor nerves are the most common cranial nerves that are involved in injury. Meningiomas occur in about 50% of patients and can be multi-tumour. They are found in the brain protrusions, the base of the skull, along the crescent of the cerebrum, and around the optic nerves. Like other tumors in NF2, they grow at different rates and have periods of relative inactivity. Meningiomas that cause cerebral edema are more severe than tumors without edema and require more rapid intervention. Headache, seizures, loss of balance, and decreased vision may occur, depending on the tumor location and the degree of compression of adjacent brain tissue. One study found that having meningiomas with NF2 increased the relative risk of death by 2.5 times compared with patients without meningiomas.
Spinal cord tumors are seen in nearly 70% of NF2 cases. Spinal cord schwannomas and extramedullary meningioma are the most common. Weakness, altered sensation, pain, and bowel/bladder disturbances can occur when these lesions cause spinal cord compression and nerve compression. Endometrial tubular tumors are seen in 33% to 53% of patients and tend to be found in the cervical spine and cervical-medullary joints. They can span multiple spinal cord segments and often contain cystic components. The majority of endothelial medullomas in NF2 have a benign course, are asymptomatic, and do not require intervention.
A distinct polyneuropathy with NF2 is a lesser-known feature of this pathology, with one study reporting clinical features of neuropathy in nearly 50% of NF2 patients. While axonal neuropathy is more common, demyelinating changes can also be found on neurotransmission. Intriguingly, polyneuropathy in NF2 occurs without accompanying schwannoma. Therefore, an NF2 patient with loss of tendon reflexes that is not explained by peripheral nerve schwannomas may need evaluation for an underlying neurological disease. Children and adults with NF2 can have a mononeuropathy, most commonly the facial nerve. In addition, a poorly understood polio-like condition leading to irreversible lower limb paralysis has been reported in pediatric NF2 and more rarely in adults.
2.3 Eyes About 80% of NF2 patients have eye damage. Subcapsular cataracts, a type of cataract, fall within the diagnostic criteria and are seen in 60% to 80% of patients. Other ocular findings include preretinal membrane, retinal extravasation, wedge opacities in children, optic disc glioma, and optic nerve sheath meningioma. Unexplained amblyopia and strabismus are well-described signs in early pediatric NF2. Continuous detection and monitoring of these abnormalities is important because they are potentially vision-threatening.

Khoảng 80% bệnh nhân bị u xơ thần kinh týp 2 gặp các vấn đề về mắt
3. Illustrated clinical case
A 10-year-old female presents to the emergency department with a 2-week history of worsening headache, nausea, and vomiting. The patient was previously diagnosed with Viral Syndrome, but his family brought him to the emergency department after he developed numbness in his face and right arm accompanied by intermittent difficulty speaking. The patient had no history of paralysis or seizures. Two years ago, the patient had surgery for congenital cataract in the right eye.
In the emergency department, neurologic examination and general examination are normal. Cranial CT showed an hyperattenuating mass in the left frontal parietal region with adjacent palatine hypertrophy and 2 mm midline deviation suggesting a meningioma. Cranial MRI showed a large, enhancing extraaxial mass in the left hemisphere with central necrosis and surrounding edema consistent with a meningioma. In addition, bilateral cranial nerve VII and VIII schwannomas were found. MRI of the spine was normal. The diagnosis of neurofibromatosis type 2 (NF2) was then confirmed.
Comment:
This case illustrates the fact that while adults with NF2 often present with symptoms associated with vestibular schwannomas, children with NF2 often present with eye symptoms, meningiomas, non-vestibular schwannomas and spinal cord tumors.
4. Management of neurofibromatosis type 2
As with many other multisystem diseases, the best management of NF2 is comprised of specialists from a variety of disciplines, including medical genetics, otolaryngology, neurosurgery, neurosurgery, oncology, and oncology. mail, ophthalmology, and audiology. Evaluation at the time of diagnosis should include neuroimaging of the brain/inner ear canal and spine with and without contrast injection, ophthalmology, neurologic examination, and audiology. Depending on the lesion and symptoms noted, follow-up periodic follow-up with neuroimaging, audiometry, and clinical evaluation is necessary along with follow-up with appropriate specialists.
Genetic testing and counseling are important components of the management of patients with NF2. There is a high prevalence of mosaicism in NF2, which can complicate the definitive diagnosis of NF2. As an autosomal dominant disease, patients with two generations of family members with a confirmed diagnosis of NF2 have a 50% risk of passing the disease on to the next generation. However, more than 50% of NF2 cases are due to new mutations without a family history, and the children of these people may have a 50% less risk of developing NF2, as some of these cases can is mosaic. Genetic studies have shown that 25% to 33% of new mutations are mosaic, with an NF2 mutation seen only in tumors but not in blood lymphocytes. A mutation can be identified in lymphocytes in only about 60% of people who meet the diagnostic criteria for NF2 without a family history.
Notably, while treatment has traditionally been focused on surgical intervention, over the past decade the focus has shifted to the development and use of chemotherapy, such as bevacizumab, that target biologic pathways related to tumor growth. These studies have therefore yielded mixed results in terms of tumor size reduction and hearing recovery, but newer drugs are being investigated. Surgery remains the cornerstone of treatment for other spinal cord and brain tumors in NF2, including schwannomas, meningiomas, and endothelial cystadenomas. Cancer intervention with chemotherapy and radiation is reserved for more malignant lesions, which are less common in NF2.
Please dial HOTLINE for more information or register for an appointment HERE. Download MyVinmec app to make appointments faster and to manage your bookings easily.
References:
Rosser T. Neurocutaneous Disorders. Continuum (Minneap Minn) 2018;24(1, Child Neurology):96-129.