This is an automatically translated article.
Article by Master, Doctor Vu Duy Dung - Department of General Internal Medicine - Vinmec Times City International Hospital
Neurofibromatosis type 1 (NF1) is the most well-understood and most common neurodermatologic disease, with an incidence of approximately 1/3000 to 1/4000 individuals. NF1 is an autosomal dominant disease caused by mutations in the NF1 gene located on chromosome 17q11.2. Approximately 50% of NF1 cases are familial, and the remainder occur sporadically.
The NF1 gene encodes the neurofibromin protein, which acts as a guanine triphosphatase (GTPase)-GAP-activating protein - GAP and has important roles in cell development and signaling pathways, participating in the downregulation of signaling pathways RAS-MAPK inhibits tumors. NF1 is a multisystem disease that affects mainly the skin, central nervous system, peripheral nervous system, eye, and musculoskeletal system.
1. Diagnosis of neurofibromatosis type 1
In 1987, the National Institutes of Health (NIH) Consensus Development Committee established the diagnostic criterion for NF1 which is still today the gold standard for clinical diagnosis. In most cases, the diagnosis of NF1 can be made on clinical grounds by a physician experienced in the application of diagnostic criteria, but genetic testing may be necessary in such cases. atypical. In addition, the clinical discovery of milder Legius syndrome (discussed in another article) has increased the need to perform genetic testing to differentiate it from NF1. Because the clinical features of NF1 evolve over time, it can also be difficult to confirm the diagnosis in young children with many coffee-milk spots and no other signs of NF1, so genetic testing is also helpful. may be appropriate in these cases. However, a definitive clinical diagnosis is possible based on the NIH diagnostic criteria for up to 75% of children up to 6 years of age and most children up to 10 years of age. Notably, some patients may have a mosaic or localized form of NF1 that involves only one part of the body and does not put them at risk for many of the medical complications that may be seen in NF1 patients. Body.
2. Clinical features of neurofibromatosis type 1
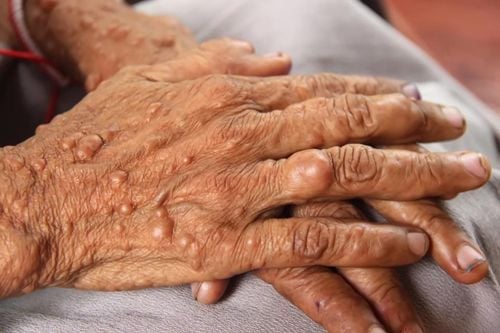
NF1 primarily affects the skin, central nervous system, peripheral nervous system, eyes, and musculoskeletal system, but rarer complications can occur in other organ systems.2.1. Skin Coffee-milk spots are the core clinical feature in NF1 and are seen in most cases. These are punctate, mottled hyperpigmented lesions that are usually present at birth and gradually increase in size and number over the first few months to years of life. Freckles accompanied by punctate, punctate hyperpigmented lesions of 1mm to 3mm in the axillary and inguinal regions are found in nearly 90% of cases. Freckles most commonly appear during the toddler years and can help confirm the diagnosis of NF1 in infants with numerous coffee-milk spots.

Các đốm cà phê sữa ở vùng nách hoặc bẹn là đặc điểm lâm sàng cốt lõi của bệnh nhân mắc u xơ thần kinh týp 1
2.2. Peripheral Nervous System Neurofibromas are benign nerve sheath tumors found along the length of the peripheral nerves. They usually appear during the pre-teen and teen years and are seen in nearly all patients by adulthood. Neurofibromas are usually classified into cutaneous and subcutaneous groups. They are unlikely to be malignant but can cause superficial disfigurement and itching.
Pusular neurofibromas occur in approximately 30% of NF1 patients and are representative of more complex schwannomas that originate in nerve fiber bundles or plexuses. They are thought to be congenital and can be found anywhere, internally or externally, on the body. When visible externally, they may have a hyperpigmented irregular surface with or without alopecia. Internal plexus neurofibromas may be visible only on imaging studies such as MRI. Neurofibromatosis plexus tumors are a significant cause of morbidity in NF1. As they progress over time, plexiform fibroids become intricately related to surrounding tissues. As such, they often lead to significant surface deformities, compression of organs, erosion of adjacent bone, vascular damage, and neurological deficits such as paralysis, sensory disturbances, and pain.
Unlike cutaneous neurofibromas, plexus neurofibromas are at risk of malignant transformation, becoming malignant peripheral nerve sheath tumors. Epidemiological studies have determined the lifetime risk of malignant transformation to be approximately 10%. Malignant peripheral nerve sheath tumors can occur sporadically in people without NF1, but they occur at a younger age and are more frequent in patients with NF1. Clinical warning signs of malignancy include rapid plexus growth, severe pain, new neurological deficits, and a change in plexus texture to a more solid mass. Malignant peripheral nerve sheath tumors present a treatment challenge because they often cannot be completely resected and are resistant to chemotherapy. The 5-year survival rate is poor, at 20% to 50%.
2.3. Central nervous system Gliomas of the visual pathway are a pediatric form, seen in 15% to 20% of children with NF1. The group under 6 years of age is most at risk for optic pathway gliomas, with a mean age of tumor detection being about 3 years. WHO grade I astrocytomas, optic pathway gliomas very often have a painless course but become symptomatic in one-third to one-half of patients. They can occur at any site of the visual pathway, including the anterior optic nerve, the optic nerve, the optic canal, and the optic ray. Several studies have demonstrated that ophthalmic and post-optical interference lesions are most likely to progress, whereas pre-optical lesions have a more benign course. Signs may include optic nerve atrophy, protrusion, pupillary abnormalities, decreased visual acuity, and decreased color vision. Additional lesions of the hypothalamus are not uncommon and can lead to endocrine disorders such as precocious puberty. Chemotherapy is usually based on progression of visual symptoms but can sometimes be indicated as lesions progress on neuroimaging.
Cranial MRI in the pediatric NF1 group also frequently shows T2 hyperintense lesions, which are now considered benign and should not be confused with brain tumors. Hyperintense lesions on T2-pulse MRI occur in 60% to 80% of pediatric NF1 patients and are found in typical locations of the brainstem, basal ganglia, thalamus, and cerebellum. They are non-enhanced and, by definition, do not cause mass effects or neurological deficits. These lesions progress and then gradually regress during childhood, with many cases spontaneously resolving in adulthood. There are some pathological studies of these interesting lesions, but they are hypothesized to represent sites of myelin edema because of their histological presentation of vacuolar or spongy tissue changes. There is a large body of literature from research efforts to determine the association between hyperintense lesions on T2 pulsed MRI and neurocognitive deficits in children with NF1. Although there is general agreement that such an association is likely, the true nature and pathogenesis are still unclear.
Neurocognitive and behavioral deficits are well-understood neurological complications of NF1. Learning disabilities are seen in 30% to 70% of children with the disease. Compared with siblings without the disease, low IQ is seen in children with NF1, but intellectual disability (IQ below 70) is uncommon. A wide spectrum of cognitive domains will be affected, including verbal memory, visual-spatial, and executive functions such as attention and working memory. In addition, approximately 30% to 50% of children with NF1 meet the diagnostic criteria for attention deficit hyperactivity disorder (ADHD), which can further impact academic and social consequences. More recent research has shown an increased prevalence of autism symptoms and impaired social functioning in children with NF1.
2.4. Eyes Lisch nodules (iridotomyomatous lesions) are observed in approximately 95% of patients over 20 years of age. Slit-lamp eye exams are often needed to see them. They are markers of NF1 but do not cause visual or other ocular symptoms.
2.5. Musculoskeletal System Specific musculoskeletal symptoms of NF1 are part of the diagnostic criteria and are often easily detected at birth. The most common findings include sphenoid dysplasia, anterior chest wall malformations (chicken and concave thorax), tibial/fibula curvature, decreased bone density, and osteoporosis. A prosthesis occurs when the long, thin bones that are easily damaged in NF1 are broken, which are often difficult to heal and require surgical fixation. Vertebral scalloped and neurofibromatosis tumors of the spine can cause dystrophic scoliosis, which presents at a young age, is more severe, and progresses more rapidly than idiopathic scoliosis. In addition, patients with NF1 have decreased bone mineral density and higher fracture rates than the general population, possibly because of alterations in osteoblast or osteoclast function in NF1.
2.6. Other organs Vascular disease is less common but is a potentially dangerous complication of NF1. The study demonstrated that aberrant neurofibromin expression and dysregulation of the RAS-MAPK pathway in vascular endothelial cells induce proliferative and angiogenic changes. Vascular abnormalities seen in NF1 patients include retinal artery stenosis, moyamoya syndrome, cerebral aneurysm, and narrow or tortuous cerebral vessels. Narrowing of the retinal arteries can lead to increased blood pressure. Recent literature has shown that, compared with the general population, rates of stroke are significantly higher in both children and adults with NF1. Adults and children are at higher risk of hemorrhagic stroke, while children are particularly at risk of ischemic stroke. Finally, because NF1 is induced by aberrations of tumor suppressor biological pathways, there is an increased incidence of brain and other tumors in NF1. Pheochromocytoma, gastrointestinal stromal tumor, leukemia, brain tumor, and breast cancer are all more common in NF1 patients than in the general population.

Bệnh lý mạch máu thường ít gặp hơn nhưng là biến chứng tiềm tàng nguy hiểm của bệnh u xơ thần kinh
3. Diagnostic criteria for neurofibromatosis type 1
The diagnostic criteria for neurofibromatosis type 1 according to the National Institutes of Health require two of the following seven clinical features:
Six or more coffee-milk spots > 5 mm in diameter in prepubertal children and > 15mm in postpubertal children Two or more neurofibromas or one plexiform neurofibroma Axillary or inguinal freckles Visual pathway glioma Two or more Lisch nodules One bone lesion isolated (long-bone thinning, sphenoid dysplasia) A first-generation relative with neurofibromatosis type 1 according to the above criteria.
4. Management
NF1 is best managed by physicians who are experienced and familiar with the disease in a multi-specialty setting with access to specialists with expertise in the management of NF1-related complications. There are published guidelines for medical monitoring recommendations for children with NF1 and are in the process of being updated, but such recommendations are lacking for adult patients with NF1.
As the signs and symptoms of NF1 evolve over time, long-term follow-up is necessary and specific management recommendations are age-related. Genetic counseling is an integral part of patient care and is most important at the time of diagnosis and with childbearing planning. During childhood, skin examination and monitoring of vision, bone growth, blood pressure, and annual neurodevelopmental monitoring are important.
Eye exams are recommended annually until age 8 and every 2 years thereafter until age 18 to evaluate glioma-associated changes of the visual pathway. Although a matter of controversy, the medical evidence does not support the use of screening brain MRI to evaluate optic pathway gliomas, but if the child has decreased visual acuity, internal abnormalities Seizures, significant headache, seizures, significant head size increase, or other neurologic symptoms of concern should warrant brain MRI. Referral to developmental specialists should be considered in children with signs of learning disability, attention deficit hyperactivity disorder, or signs of autism.
Management of adults with NF1 focuses on monitoring for benign, malignant tumors, bone conditions, and vascular disease as well as the progression of any complications seen early in life. A low threshold is required when evaluating any NF1 patient for stroke if new neurological deficits are present.
As our understanding of neurofibromin functions and the RAS-MAPK pathway has expanded, biologic targeted therapies have focused on treating the most severe complications of NF1. Numerous phase 1 and phase 2 clinical trials have been performed in the treatment of optic pathway gliomas, plexiform neurofibromas, and malignant peripheral nerve sheath tumors. , and cognitive deficits associated with NF1.
Please dial HOTLINE for more information or register for an appointment HERE. Download MyVinmec app to make appointments faster and to manage your bookings easily.
References:
Rosser T. Neurocutaneous Disorders. Continuum (Minneap Minn) 2018;24(1, Child Neurology):96-129.