This is an automatically translated article.
Posted by Master, Doctor Dao Duc Dung - Specialist in Gastroenterology - Hepatobiliary tract - General Surgery - Vinmec Times City International HospitalCongenital biliary atresia is the most common cause of biliary obstruction in nursing infants. The incidence is about 1 in 10,000 live births (1/8,000 to 1/14,000). Female/male ratio = 1:0.64. The problem of diagnosis and treatment is very complicated, without surgery, the patient will die before 2 years of age due to liver failure and complications of cirrhosis.
1. Causes and pathogenesis
Biliary atresia is considered as a consequence of the ductal development of the biliary tract during the fetal period. To date, although many mechanisms have been proposed to explain the causes of biliary atresia, these mechanisms are all hypothetical:
Recanalization of the biliary tract does not occur. Biliary atresia is associated with teratogenicity during fetal development because 10-20% of cases of biliary atresia are associated with a complex malformation known as polysplenic syndrome. In addition to polysplenia, this syndrome also has one or more of the following malformations: absence of inferior vena cava, inversion of abdominal viscera, rotator cuff malformation of intestine, only two lobes on each lung, and heart defects. Biliary atresia due to viral infection in the perinatal period causes biliary obstruction that develops after inflammation. Many viruses such as hepatitis B virus, Rubella virus, CMV virus (Cytomegalo virus), EBV (Epstein Bar virus), Renovirus. In addition, biliary atrophy may be the result of an injury (toxin or virus) that causes the biliary epithelium to degrade and become primordial on the surface of the biliary cells. These antigens are recognized by circulating T cells in the blood, and when a cell-mediated immune response is initiated, the bile ducts become fibrosis and eventually atrophy.
2. Pathology
Many histopathological studies show diffuse inflammation, leading to fibrosis and disappearance of the ductal structure of the biliary tree.
However, this sclerosing state varies from patient to patient. This is the factor that explains the success of Kasai surgery in only a few patients.
Cholestasis, trabecular torsion, focal hepatocellular necrosis and central fibrosis are characteristic features of congenital biliary atrophy.
3. Classification
There are many ways to differentiate congenital biliary atresia. Macroscopically and with contrast-enhanced cholangiography, it can be classified into 3 main types:
Type I: Atrophy of the common bile duct (5%) Type II: Atrophy up to the common hepatic duct (2%). Type III: Atrophy of the entire extrahepatic biliary tract (>90%).
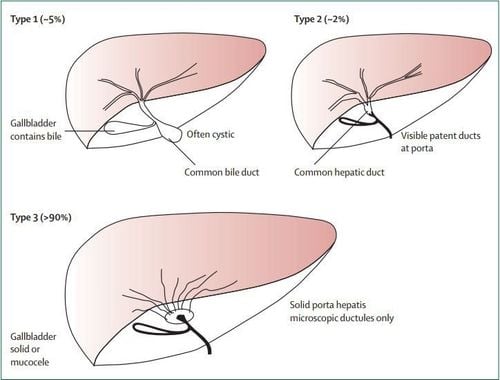
Một số dạng teo đường mật bẩm sinh trên lâm sàng
4. Symptoms of congenital biliary atresia
Symptoms of congenital biliary atresia are as follows:
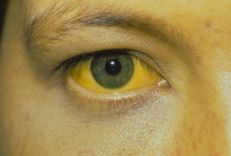
4.1. Clinical clinical presentation is mainly manifested by the classic triad of jaundice, discolored stools and hepatomegaly. Jaundice can appear right from the postpartum period, immediately after the physiological jaundice. However, in some children, jaundice appears later in life. When the baby's jaundice is over 2 weeks, it should not be considered as physiological jaundice but must quickly do exploratory tests to diagnose the cause, in which biliary atresia is the most common cause. Jaundice is often accompanied by dark urine.
Symptoms of discolored stools appear late. In most children, the meconium is normal in color. In more than half of the pediatric patients, meconium was found to be yellow or pale yellow. Discoloration of stools, rather than white stools after normal meconium, is a very valuable sign to distinguish biliary atresia from hepatitis.
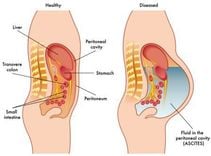
Enlarged liver is caused by cholestasis, this condition is quite likely to occur in the early stages. Firm liver with enlarged spleen, fluid in the abdomen, collateral circulation is seen in the end stage.
It is also worth noting that the majority of children with congenital biliary atresia have completely normal weight and physical growth in the first months, even until surgery. However, there are still a small number of children with malnutrition, anemia or growth retardation.
Accompanying symptoms such as intracranial bleeding, subcutaneous bleeding... are manifestations of decreased prothrombin ratio due to vitamin K malabsorption.
MORE: Distinguishing congenital biliary atresia from congenital jaundice management, anemia, hepatitis
4.2. Laboratory tests to assess biliary obstruction and liver function:
Increased blood bilirubin, mainly direct bilirubin. Alkaline phosphatase (ALP) is elevated. Prothrombin decreased. Transaminases are elevated. Tests with diagnostic value:
Ultrasound
So far, hepatobiliary ultrasound is still considered the first used imaging modality in a pediatric patient with suspected congenital biliary atresia. On hepatobiliary sonography, one looks first for the presence of the gallbladder.
Ultrasound needs to be done at two times, when the baby is hungry and after the baby is fed. The purpose of two-time ultrasound is that if the gallbladder is present and the biliary tract is not atrophied, then after feeding, the gallbladder will increase in contraction according to the normal physiological response. In addition, an important point in the diagnosis of biliary atresia with cyst formation in the hilum is that ultrasonography is useful in detecting cysts.
In addition, recently according to some authors, there is a very characteristic picture for congenital extrahepatic biliary atresia. That is what is known as the triangular cord sign. With this finding, ultrasonography allows the diagnosis of congenital biliary atresia with sensitivity of 84% and specificity of 98%.
However, ultrasound only allows suspicion but cannot confirm the diagnosis. The sensitivity and specificity of ultrasound in the diagnosis of congenital biliary atresia are not high, according to most studies, only in the range of 80-85%. In addition, in cases where a gallbladder is present, it is also not possible to distinguish between a patient with terminal atrophy of the common bile duct and neonatal hepatitis.
Phương pháp siêu âm giúp bác sĩ chẩn đoán bệnh lý teo đường mật bẩm sinh
Radioisotope hepatography (Technetium-99m)
Using radioisotope Tc-99 is widely used in European countries. The presence of radioisotopes in the small intestine after drug administration allows the exclusion of congenital biliary atresia. Normally, this radioactive substance is absorbed by liver cells and excreted through the bile into the intestine. In biliary atresia, the radioisotope is absorbed in the liver but not excreted in the intestine.
However, in cases of solid biliary syndrome or in cases of biliary hypoplasia, this imaging test is of little help in the differential diagnosis.
According to some reports, radioisotope scan for diagnosis of biliary atresia has 100% sensitivity, 87.5% specificity and 90.5% accuracy.
Endoscopic retrograde cholangiopancreatography (ERCP)
With the advent of age-appropriate endoscopic instruments for infants, through endoscopic retrograde cholangiopancreatography, it is possible to inject contrast Biliary radiograph to diagnose biliary atresia or not? According to many studies, with the advent of modern and suitable sized vehicles today, ERCP allows accurate diagnosis in 86% of cases.
The role of laparoscopy in the diagnosis of congenital biliary atresia
Today, with the rapid development of laparoscopy, many studies have begun to apply laparoscopy in the diagnosis of atrophy. extrahepatic bile, but only in a limited number of pediatric patients. Endoscopy is aimed at determining whether or not a gallbladder is present and, in combination with intraoperative cholangiography, allows the diagnosis of extrahepatic congenital biliary atresia in more than 90% of cases.
Biopsy
Biopsy is still considered a decisive factor in the diagnosis of difficult cases of congenital biliary atresia. Biopsy is often indicated in pediatric patients with gallbladder. Biopsies are performed through the common incision, laparoscopically, or even percutaneously.
The pathological features of congenital biliary atresia include:
Macroscopically: dark green cholestatic liver, rough and rough surface. Microscopically: cholestasis, fibrosis around the portal space, twisting of the liver trabes.
5. Diagnosis of congenital biliary atresia
The diagnostic methods of congenital biliary atresia are as follows:
5.1. Clinical diagnosis: Persistent neonatal jaundice + pale stools + hepatomegaly. Biochemistry: Increased serum bilirubin, GGT and blood ALP levels. Pictures: Ultrasound, radioisotope hepatography, laparoscopy combined with intraoperative cholangiography. Liver biopsy: Cholestasis, fibrosis around the portal space. 5.2. Differential Diagnosis Physiological Jaundice. Neonatal hepatitis. Congenital biliary syndrome or congenital biliary hypoplasia. Other causes of medical jaundice. In general, the diagnosis should be made as soon as possible, because the 8-week period is considered a golden age in the treatment of biliary atresia. Patients operated before 8 weeks had a much better prognosis than patients operated after 8 weeks. Therefore, in order to be able to operate the patient early, in cases of difficult diagnosis, the Japanese authors advocate using exploratory surgery.
SEE ALSO: Congenital biliary atresia: Intractable disease

Teo đường mật bẩm sinh cần được chẩn đoán phân biệt với một số loại vàng da khác
6. Treatment of congenital biliary atresia
The method to treat congenital biliary atresia is surgery required. This is the only and first method for young children with this disease.
6.1. Preparing the patient before surgery Vitamin K is given through the system at a dose of 1-2mg/kg/day in the first days waiting for surgery.
Fasting 24 hours before surgery and correcting nutritional and laboratory disorders.
6.2. Indications: Previously, most authors believed that surgery could only be successful in pediatric patients who were operated before the 4th month, that is, before 90 days of age. Recently, however, some studies have shown that success rates are still high in pediatric patients who are operated on later, even up to 120 days of age. Therefore, surgery is still indicated in late-diagnosed pediatric patients, especially those diagnosed between 90-120 days of age.
Surgery to connect the common hepatic duct - jejunum: Applied for the so-called curable. Dissection of the hilum of the liver may reveal a cystic structure that, when transversely, shows bile leakage. This cyst was not removed but had to be used for anastomosis. Kasai surgery: Also known as porto-jejunostomy, was performed in 1955 by Morio Kasai (a Japanese surgeon) and later became the standard surgery to treat Treatment of cases of total atrophy of the extrahepatic biliary tree or atrophy of the common hepatic duct - the so-called incurable forms of congenital biliary atresia. The principle of surgery according to the Kasai method is to resect the entire extrahepatic biliary tract in a single mass, followed by creating a biliary-digestive communication using a loop of small intestine to connect with the hilum of the liver. In this, this communication is ensured by the existence of microscopic biliary structures found at the section of the hepatic hilum.
6.3. Complications and postoperative care Care and monitoring of complications after surgery for congenital biliary atresia are as follows:
Postoperative care:
Antibiotics: use broad-spectrum antibiotics that have good effects on Gram-negative bacteria . Corticosteroids: Aim to limit the inflammatory response as well as increase the bile flow, but the opinion is still not completely unified among the authors. Diuretic drugs (UDCA - ursodeoxycholic acid): As with the use of corticosteroids, opinions are still divided. Many authors use UDCA for the purpose of having both biliary and hepatoprotective effects. In addition to the above care methods, parents also need to pay attention to the nutrition after treatment for congenital biliary atresia so that the child can recover quickly.
Complications after surgery:
Early complications: The development of biliary atresia after surgery is often complicated and heavy. In addition to the problem of anastomosis, which is a possible complication, two other problems can also occur: Abdominal bleeding and Electrolyte disturbances (mainly hyponatremia). Inflammation and infection of the biliary tract: Rates vary by study and range from 40-60%. The origin may be from retrograde infection following biliary-enteric anastomosis. The clinical presentation is in the form of fever, decreased bile outflow and increased bilirubin in the blood. These pediatric patients must be treated with fluids and antibiotics. Usually, after 6-9 months after surgery, when the flow and secretion of bile have reached normal levels, infection as well as cholangitis will decrease. Portal hypertension (ALTMC): At the time of surgery, more or less, all pediatric patients had some degree of cirrhosis. Increased ALTMC appeared after surgery with a frequency of about 34-76%. Gastrointestinal bleeding due to rupture of esophageal varices may occur in 20-60% of pediatric patients after surgery. Accordingly, conservative treatment is recommended because the tendency to bleed from ruptured veins decreases with age. If conservative treatments fail, surgery for portal-host drainage or liver transplantation is required. Other complications may be encountered after surgery such as: metabolic disorders, decreased liver function, malnutrition, cysts in the liver (bile duct cysts), hepatopulmonary syndrome, malignant liver disease.
MORE: Can congenital biliary atresia be treated with stem cell transplantation?

Nhiễm trùng đường mật có thể xảy ra sau mổ teo đường mật bẩm sinh
7. Liver transplant problem
Neonates who do not clear jaundice after Kasai surgery and those who develop complications or end-stage chronic liver disease despite initial successful Kasai surgery are all cases requiring liver transplantation. Congenital biliary atresia is the most common indication for liver transplantation in children. Most of these cases require a liver transplant within the first few years of life.
The timing of liver transplantation depends on liver function, nutritional status, symptoms and the occurrence of complications. Hepatic arterial hypertension through the resistance index (RI) measured on Doppler ultrasonography is an ominous sign and an indication of the need for acute liver transplantation. However, up to 20% of patients after Kasai surgery will have good liver function results and live to adulthood without a liver transplant.
The 5-year survival rate after liver transplantation for biliary atresia is about 90%, the 10-year survival rate has been reported to be about 81% and current technology will improve this outcome. The current big problem is the shortage of transplant organs and the high cost of liver transplantation. To solve the problem of shortage of transplanted organs, the development of liver transplantation techniques such as: Liver division for transplantation (with brain-dead donors) and liver transplantation from living donors (or to put it in plain English). is a liver transplant for a child by taking a part of the liver of a parent or a relative to transplant to a child) has markedly reduced the mortality rate on the transplant waiting list. The combination of Kasai surgery and liver transplantation helped change a disease that was almost certainly fatal in the 1960s into one with a five-year survival rate of about 90%. Furthermore, long-term studies have shown a link between markedly improved quality of life in patients after Kasai surgery alone and after liver transplantation.
Along with the development of modern medicine, early diagnosis of biliary atresia and minimally invasive surgery increase the success rate of Kasai surgery. Kasai surgery helps to delay or reduce the need for a liver transplant. The development of liver transplant centers with the application of modern liver transplantation techniques, the quality of post-transplant care and the development of new immunosuppressive drugs helps to increase survival rates as well as quality of life after transplantation. graft.
Congenital biliary atresia is a dangerous disease but it is easily confused with many other congenital diseases. Therefore, in order to diagnose this disease early, when parents see that their child has signs of prolonged pathological jaundice, it is necessary to take the child to a specialized medical facility for early examination, identification and treatment.
Vinmec International General Hospital is the address that has successfully treated congenital biliary atresia by liver transplantation and stem cell transplantation. With the "golden hand in pediatric surgery" of Professor, Dr. Nguyen Thanh Liem - the first person to introduce hematopoietic stem cell transplantation to treat diseases in our country, has helped many children with biliary atresia. Congenital has a good prognosis later in life. Therefore, if the child shows signs of congenital biliary atresia, jaundice, hepatomegaly, splenomegaly and other abnormal signs, parents can take the child to Vinmec International General Hospital for examination and treatment. Get the best support and advice from doctors and health professionals.
Please dial HOTLINE for more information or register for an appointment HERE. Download MyVinmec app to make appointments faster and to manage your bookings easily.