Bài viết được viết bởi BS Đỗ Phước Huy - Trung tâm Công nghệ cao Vinmec
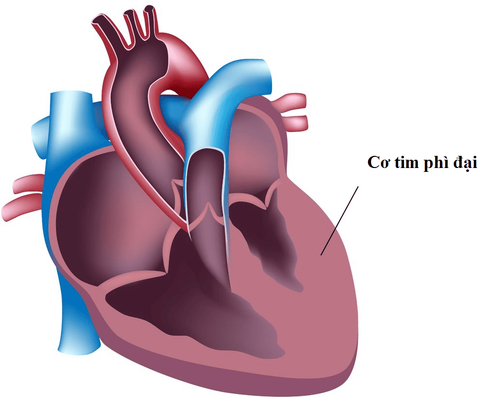
Rối loạn dự trữ glycogen nhóm 2 (GSD 2) (OMIM #232300), hay còn gọi là bệnh Pompe, là một rối loạn di truyền gây ra do sự thiếu hụt enzyme tham gia trong quá trình chuyển hóa glycogen. Rối loạn dự trữ glycogen nhóm 2 lần đầu tiên được mô tả bởi nhà giải phẫu bệnh Joannes Cassianus Pompe vào năm 1932 trên một bé gái 7 tháng tuổi chết vì cơ tim phì đại.
1. Thông tin chung
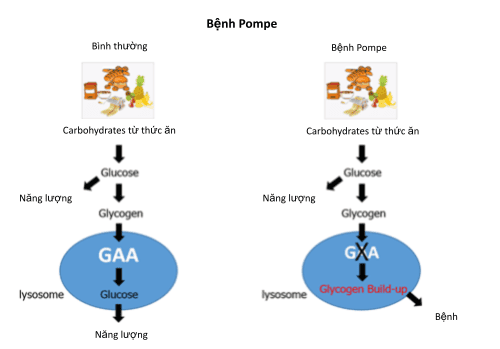
Rối loạn dự trữ glycogen nhóm 2 do sự khiếm khuyết enzyme acid alpha-glucosidae, hệ quả glycogen không được biến đổi thành glucose và tích trữ trong lysosome (một bào quan chứa các enzyme liên quan quá trình tiêu hóa các chất trong tế bào).
Bệnh nhân rối loạn dự trữ glycogen nhóm 2 có 2 thể (thể khởi phát sớm – Infantile Onset Pompe Disease và thể khởi phát muộn – Late Onset Pompe Disease) với biểu hiện lâm sàng rất biến đổi từ triệu chứng cho đến tuổi khởi phát và tiên lượng.
2. Lâm sàng của rối loạn dự trữ glycogen type 2
2.1 Thể khởi phát sớm (IOPD)
Rối loạn dự trữ glycogen nhóm 2 thể khởi phát sớm thường được định nghĩa là khởi phát trước 12 tháng tuổi và có kèm bệnh lý cơ tim. Tuy nhiên trên thực tế lâm sàng, triệu chứng thường rõ rệt vào khoảng 4 tháng tuổi. Bên cạnh đó có những ca lâm sàng được báo cáo với triệu chứng xuất hiện từ lúc còn bào thai. Các triệu chứng lâm sàng thường gặp gồm yếu cơ, bệnh lý cơ tim, tim và gan to, khó cho trẻ bú, suy hô hấp, và nặng nề nhất là tử vong. Ngoài ra nghe kém cho đến mất thính lực cũng hay gặp và thường do cả nguyên nhân ốc tai cũng như dẫn truyền.
Bảng 1: Các triệu chứng thường gặp của IOPD
| Triệu chứng lâm sàng | Tỷ lệ thường gặp |
| Yếu cơ | 52%-96% |
| Tim to | 92%-100% |
| Gan to | 29%-90% |
| Phì đại thất trái | 83%-100% |
| Bệnh cơ tim | 88% |
| Suy hô hấp | 41%-78% |
| Phì đại lưỡi | 29%-62% |
| Khó cho bú | 57% |
| Chậm phát triển | 53% |
| Mất phản xạ gân xương | 33%-35% |
Nếu không được điều trị sớm trẻ thường chết trong khoảng 2 năm đầu đời.
2.2 Thể khởi phát muộn (LOPD)
Rối loạn dự trữ glycogen nhóm 2 thể khởi phát muộn được định nghĩa là (1) khởi phát trước 12 tháng tuổi nhưng không kèm bệnh cơ tim và (2) khởi phát sau 12 tháng tuổi.
LOPD có thể biểu hiện ở bất cứ tuổi nào với yếu cơ và suy hô hấp. Tiên lượng và tiến triển của bệnh phụ thuộc nhiều vào tuổi khởi phát, khởi phát càng sớm tiên lượng càng xấu. Với nhóm LOPD, bất thường về tim hiếm gặp, tuy nhiên các bệnh lý mạch máu thường gặp hơn ví dụ như dãn động mạch chủ xuống. Yếu cơ xương thường diễn tiến chậm hơn so với nhóm IOPD, khởi phát có thể là trẻ khó tham gia các môn thể thao, yếu cơ khi lên cầu thang... Bên cạnh yếu nhóm cơ liên quan vận động, yếu cơ hô hấp thường gặp và là nguyên nhân gây tử vong ở bệnh nhân LOPD.
LOPD thường khởi phát ở lứa trẻ lớn, tuy nhiên cũng đã có ghi nhận những trường hợp khởi phát tuổi trưởng thành. Trong những trường hợp này triệu chứng đã xuất hiện lúc nhỏ nhưng rất mơ hồ.
Biểu hiện lâm sàng chính của nhóm LOPD:
- Yếu cơ gốc chi tiến triển (gặp ở 95% số trường hợp)
- Suy hô hấp
- Giảm hoạt động thể lực
- Khó thở khi gắng sức

3. Xét nghiệm di truyền cho nhóm rối loạn dự trữ glycogen type 2
Tính tới thời điểm hiện tại, hầu hết các đột biến gen GAA được báo cáo là gây bệnh hoặc có thể gây bệnh trên ClinVar đều nằm trên vùng exon hoặc vùng splicing. Với các xét nghiệm gen khảo sát mức độ exon và vùng gần exon có thể phát hiện được các đột biến gen liên quan nhóm rối loạn dự trữ glycogen nhóm 2 này. Ngoài ra có khoảng 5% - 13% đột biến liên quan những mất lặp đoạn gen GAA gây ra rối loạn dự trữ glycogen nhóm 2 và cần thực hiện các xét nghiệm như quantitative PCR, multiplex ligation-dependent probe amplification (MLPA), and chromosomal microarray (CMA).
Hiện nay xét nghiệm giải trình tự gen thế hệ mới (NGS) cho phép khảo sát nhiều gen và nhiều vùng của gen một lúc. Trong bệnh lý rối loạn dự trữ glycogen nhóm 2, do chủ yếu đột biến xảy ra vùng mã hóa (exon) và vùng nối (splicing site) nên các xét nghiệm giải trình tự gen vùng mã hóa như WES có thể đáp ứng được nhu cầu này, cũng như giúp chẩn đoán phân biệt với các nhóm bệnh lý khác trong nhóm rối loạn dự trữ glycogen.
4. Điều trị với nhóm rối loạn dự trữ glycogen type 2
Điều trị trong nhóm rối loạn dự trữ glycogen nhóm 2 bên cạnh điều trị triệu chứng, việc điều trị liệu pháp thay thế enzyme (ERT – Enzyme replacement therapy) cần được đặt ra ngay khi có chẩn đoán nhằm giảm thiểu các biến chứng. Tuy nhiên để biết được trẻ có phù hợp với ERT hay không cần xác định trạng thái miễn dịch chéo (cross-reactive immunologic material – CRIM). Có hai cách xác định yếu tố CRIM:
- Định lượng protein axit alpha-glucosidase được thực hiện bằng phương pháp dựa trên kháng thể trong nguyên bào sợi;
- Xét nghiệm giải trình tự gen GAA để xác định xem các biến thể gây bệnh có dẫn đến mất hoạt tính của enzyme GAA (tức là CRIM âm tính)
Và hiện nay Sanofi Genzyme có chương trình hỗ trợ điều trị cho các trẻ đã có chẩn đoán rối loạn chuyển hóa glycogen nhóm 2 phù hợp với liệu pháp thay thế enzyme.
Để đặt lịch khám tại viện, Quý khách vui lòng bấm số HOTLINE hoặc đặt lịch trực tiếp TẠI ĐÂY. Tải và đặt lịch khám tự động trên ứng dụng MyVinmec để quản lý, theo dõi lịch và đặt hẹn mọi lúc mọi nơi ngay trên ứng dụng.