This is an automatically translated article.
The article was professionally consulted by Specialist Doctor I Nguyen Thi My Linh - Neonatologist - Department of Pediatrics - Neonatology - Vinmec Danang International General Hospital.Smith-Magenis syndrome (SMS) is a complex developmental disorder that results in multiple organ systems being affected, characterized by a pattern of abnormalities at birth (congenital) ) as well as behavioral and cognitive problems.
1. What is Smith-Magenis syndrome?
Smith-Magenis syndrome is a birth defect caused by a microscopic deceleration or abnormality of chromosome 17. Key features of Smith-Magenis Syndrome (SMS) include mild to moderate intellectual disability , delays in speech and language skills, facial features, sleep disturbances, and behavioral problems. Smith-Magenis syndrome affects at least 1 person in 25,000 people worldwide.Smith-Magenis syndrome is a chromosomal condition involving chromosome 17. Most people with SMS have deletions of genetic material from a specific region of chromosome 17 (17p11.2). . Although this region contains many genes, researchers have recently discovered that the loss of a specific gene that retinoic acid 1 or RAI1 causes is responsible for most of the characteristic expressions of retinoic acid. This situation. In addition, other genes in chromosome 17 contribute to the variability and severity of clinical features. The loss of other genes in the deleted region may help explain why features of Smith-Magenis syndrome vary among affected individuals. A small percentage of people with Smith-Magenis syndrome have a mutation in the RAI1 gene instead of a chromosomal deletion.
These deletions and mutations lead to the production of an abnormal or non-functional version of the RAI1 protein. RAI1 is a transcription factor involved in the communication messages between DNA and RNA.

Hội chứng Smith-Magenis là một khuyết tật bẩm sinh liên quan đến nhiễm sắc thể 17
2. Signs to recognize Smith-Magenis . syndrome
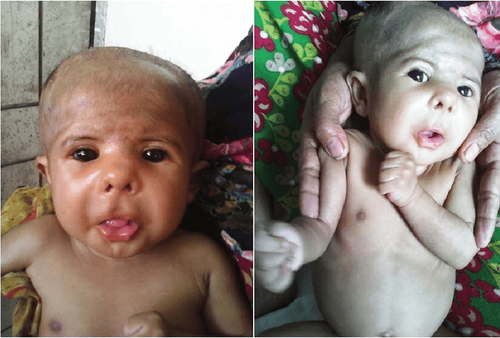
People with Smith-Magenis Syndrome are characterized by a short flat head, prominent forehead, wide square face, deep eyes, underdeveloped face, wide nose bridge, short nose, upturned upper lip, and small chin since childhood. small and become relatively more prominent for their age. These facial differences may be less obvious in childhood, but they often become more distinctive as the child matures. Dental abnormalities are also common.Children with Smith-Magenis syndrome in infancy often have problems with suckling, not growing like their peers, weak muscle tone, prolonged napping. Later in adulthood, children will experience sleep disturbances (difficulty falling asleep, frequent awakenings throughout the night, and daytime sleepiness), as well as behavioral problems.
These behavior problems can manifest in a variety of ways, such as frequent tantrums, impulsivity, anxiety, distraction, aggression, and self-injurious behaviors including self-beating, self-biting and self-splitting.
In addition to the defiant behaviors described above, children with Smith-Magenis syndrome may also exhibit stereotypical behaviors including hand-holding, object-holding, hand-to-mouth, teeth grinding, and muscle swinging. body and rotate or the object rotates.
Other signs and symptoms of Smith-Magenis syndrome include short stature, abnormal curvature of the spine (scoliosis), decreased sensitivity to pain and temperature, and possibly difficulty going to the bathroom towel and hoarse voice. Some patients also develop ear abnormalities that lead to hearing loss and possibly vision problems. Although less common, heart and kidney defects have also been reported in people with Smith-Magenis syndrome.

Trẻ sơ sinh mắc hội chứng Smith-Magenis thường không phát triển như bạn bè
3. Diagnosis and treatment of Smith-Magenis . syndrome
The diagnosis of Smith-Magenis Syndrome (SMS) is usually made through a clinical blood test called a chromosome analysis. Diagnosis can also be made through cytogenetic testing and FISH (fluorescence in situ hybridization) or by chromosomal microarray (CGH) analysis.Most people diagnosed with SMS are born with a small deletion of the 17th pair of chromosomes in the body. It is this deficiency, known as 17p11.2, that is responsible for the SMS conditions.
Genes commonly deleted in people with SMS have been narrowed down to a "critical region" consisting of about 25 genes. All deletions included deletions of the RAI1 gene.
In cases where deletions are not detected, sequencing of the RAI1 gene should be considered. About 90% of SMS cases are due to deletions, the remaining 10% are due to mutations occurring in the RAI1 gene.
Mutations in RAI1 can be identified by sequencing of the RAI1 gene. RAI1 sequencing can be done using different types of genetic tests. Trials include RAI1 sequencing:
Whole Exome Sequencing: This test sequencing all of an individual's genes Genetic testing via console: This test includes sequencing only self for some selected genes. RAI1 must be listed as a gene in the table. Panels that treat epilepsy, autism spectrum disorders, and/or intellectual disability often include RAI1 Sequence RAI1: This test only sequenced RAI1.

Trình tự RAI1 có thể được xác định bằng các loại xét nghiệm di truyền
In addition, patients and families should consult genetic counselors during treatment. There is currently no clear treatment for patients with Smith-Magenis syndrome other than supportive therapy and symptomatic relief. Early diagnosis and treatment is essential to help children become independent as soon as possible.
Special education with speech and speech therapy, physical therapy, and occupational therapy is needed for children with Smith-Magenis syndrome to help children become independent. Sensory integration therapy will help children respond better to stimuli because children with this syndrome respond very poorly.
Medicines can be used to treat behavioral and attention-seeking problems in children with Smith-Magenis syndrome. It is advisable to consult a gastroenterologist in conjunction with a pediatrician for children with feeding difficulties. Your doctor may prescribe anticonvulsants to control seizures in cases where your child has epilepsy.
Any questions that need to be answered by a specialist doctor as well as customers wishing to be examined and treated at Vinmec International General Hospital, please contact the Website for the best service.
Please dial HOTLINE for more information or register for an appointment HERE. Download MyVinmec app to make appointments faster and to manage your bookings easily.