Bài viết được viết bởi Bác sĩ Đỗ Phước Huy, Trung tâm Công nghệ cao Vinmec
Rối loạn dự trữ glycogen (Glycogen Storage Diseases – GSDs) là một nhóm rối loạn chuyển hóa di truyền do bất thường một trong nhiều enzyme tham gia trong quá trình sinh tổng hợp và phân giải glycogen.
1. Tổng quan về rối loạn dự trữ glycogen
Rối loạn dự trữ glycogen thường được chia làm ba nhóm tùy thuộc cơ quan bị ảnh hưởng gồm nhóm triệu chứng chính liên quan đến gan, nhóm biểu hiện bệnh lý thần kinh cơ và nhóm biểu hiện cả hai. Mức độ nặng của biểu hiện bệnh (từ nhẹ cho đến tử vong), thời gian khởi phát (trước sinh, sơ sinh, trẻ nhỏ hay trẻ lớn), và tiên lượng phụ thuộc rất nhiều vào nhóm Rối loạn dự trữ glycogen, loại đột biến, thời điểm bắt đầu điều trị...
Để chẩn đoán nhóm rối loạn dự trữ glycogen, cần kết hợp cả triệu chứng lâm sàng, các kết quả xét nghiệm sinh hóa, mô bệnh học và xét nghiệm di truyền. Trẻ có thể cần sinh thiết gan, cơ cũng như kết hợp với xác định hoạt độ enzyme. Tùy thuộc vào từng nhóm rối loạn dự trữ glycogen khác nhau mà sự thiếu hụt enzyme biểu hiện chủ yếu ở gan, cơ, nguyên bào sợi da, và hiếm khi phát hiện ở các tế bào máu. Tuy nhiên, nếu xác định được nhóm rối loạn dự trữ glycogen chủ yếu biểu hiện ở bất thường gan hay cơ sẽ giúp định hướng trong quá trình chẩn đoán, tăng khả năng chẩn đoán với sinh thiết gan hay cơ và hoạt độ enzyme.
Xét nghiệm giải trình tự cũng như sự ra đời của giải trình tự gen thế hệ mới (NGS – Next generation sequencing) đang dần trở thành phương pháp chủ yếu để chẩn đoán nhóm rối loạn dự trữ glycogen. Từ một xét nghiệm có vai trò như kiểm chứng lại các trường hợp hoạt độ enzyme không rõ ràng, giải trình tự gen đang dần trở thành tiêu chuẩn vàng trong chẩn đoán xác định. Những lợi ích của xét nghiệm giải trình tự gen gồm:
- Giảm tỷ lệ chẩn đoán xâm lấn bằng sinh thiết.
- Với hoạt độ enzyme cần có một lượng mẫu mô tươi nhất định (thông qua sinh thiết), giải trình tự gen có thể thực hiện thông qua mẫu máu, mẫu mô, mẫu phết niêm mạc hay mẫu nước bọt (không phổ biến).
- Khi biết được đột biến, có thể tiến hành chẩn đoán tiền sinh, cũng như tiên lượng cho trẻ hiện tại và/hoặc thai kỳ tiếp theo.
2. Quá trình chuyển hóa glycogen và cân bằng glucose trong cơ thể
Glucose là nguồn năng lượng chính của não và nhiều cơ quan quang trọng trong cơ thể. Vì thế cơ thể cần duy trì ổn định nguồn glucose cung cấp cho não bộ, nói cách khác cần đảm bảo duy trì ổn định nồng độ glucose máu.
Để duy trì ổn định nồng độ glucose máu cơ thể sẽ tiến hành đơn lẻ hay đồng thời gồm: thông qua bữa ăn, sinh tổng hợp hay phân giải glycogen, và tiến trình cơ thể tự tạo glucose hay còn gọi là gluconeogenesis. Bởi vì chúng ta không thể liên tục ăn cũng như tiến trình gluconeogenesis cũng không thể diễn ra nhanh chóng vì thế glycogen được xem là dạng chính để dự trữ và cung cấp nhanh chóng khi cơ thể có nhu cầu.
Khi không có glucose trong chế độ ăn, hay cơ thể cần huy động glucose; glycogen ở gan nhanh chóng bị phân hủy thành glucose và được giải phóng vào máu; tương tự, glycogen trong các tế bào cơ bị thoái hóa và được sử dụng để tạo ra ATP cung cấp năng lượng cho quá trình co cơ. Khi nguồn dự trữ glycogen cạn kiệt, quá trình tạo gluconeogenesis có thể xảy ra trong các mô cụ thể, cho phép tổng hợp glucose de novo bằng cách sử dụng các chất nguồn gốc không phải carbohydrates như các amino acids...
Rối loạn dự trữ glycogen do bất thường một hay nhiều enzyme tham gia trong quá trình sinh tổng hợp và phân giải glycogen. Hệ quả gồm gan to, xơ hóa gan, hạ đường huyết, toan chuyển hóa, bất thường hoạt động của cơ như cơ vân gây yếu cơ, biểu hiện thần kinh cơ, hay cơ tim gây phì đại cơ tim...

3. Phân nhóm rối loạn dự trữ glycogen
Hiện nay có 15 nhóm và phân nhóm rối loạn dự trữ glycogen (bảng 1) với biểu hiện và tiên lượng khác nhau.
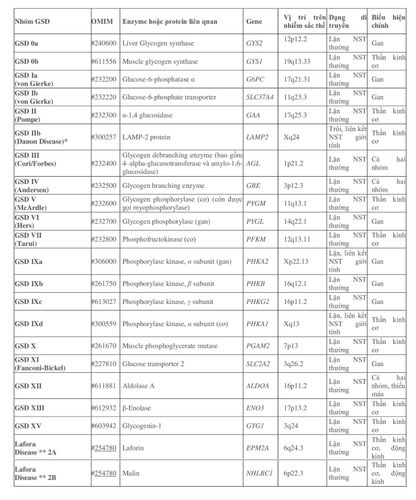
*Bệnh Danon (Danon Disease) từng được phân loại thành một phân nhóm của bệnh Pompe với nồng độ acid maltase hay alpha-glucosidase bình thường (Danon và công sự, 1981). Tuy nhiên theo Nishino và cộng sự, 2000 phân loại thành pseudoglycogenosis II do không phải luôn luôn có sự tăng glycogen.
** Bệnh Lafora có một số ý kiến phân loại vào nhóm rối loạn dự trữ glycogen do liên quan đến quá trình chuyển hóa và tích trữ glycogen tại não tạo thể Lafora (Lafora bodies)
Để đặt lịch khám tại viện, Quý khách vui lòng bấm số HOTLINE hoặc đặt lịch trực tiếp TẠI ĐÂY. Tải và đặt lịch khám tự động trên ứng dụng MyVinmec để quản lý, theo dõi lịch và đặt hẹn mọi lúc mọi nơi ngay trên ứng dụng.
Bài viết tham khảo nguồn:
https://www.omim.org/
https://medlineplus.gov/genetics/condition/
Ellingwood, S. S., & Cheng, A. (2018). Biochemical and clinical aspects of glycogen storage diseases. Journal of Endocrinology, 238(3), R131–R141. doi:10.1530/joe-18-0120
Chen, M. A., & Weinstein, D. A. (2016). Glycogen storage diseases: Diagnosis, treatment and outcome. Translational Science of Rare Diseases, 1(1), 45–72. doi:10.3233/trd-160006
Kishnani, P.S., Goldstein, J., Austin, S.L. et al. Diagnosis and management of glycogen storage diseases type VI and IX: a clinical practice resource of the American College of Medical Genetics and Genomics (ACMG). Genet Med 21, 772–789 (2019). https://doi.org/10.1038/s41436-018-0364-2
Nitschke, F., Ahonen, S.J., Nitschke, S. et al. Lafora disease — from pathogenesis to treatment strategies. Nat Rev Neurol 14, 606–617 (2018). https://doi.org/10.1038/s41582-018-0057-0