This is an automatically translated article.
Post by Master, Doctor Phan Truc - Internal Medicine Oncologist - Hematology - Oncology Center - Radiation Therapy - Vinmec Times City International Hospital
Initiation and progression of acute lymphoblastic leukemia (ALL) stem from successful mutations that induce changes in cellular function, including increased self-renewal capacity, and escape from normal proliferative control. , stops differentiation and increases resistance to cell death signaling (apoptosis). Disorders related to DNA repair may play an important role. Environmental factors such as ionizing radiation and carcinogenic chemicals are clearly associated with some patients with ALL. In most cases, however, no causative factor is evident. According to resonance theory, the process of leukaemia reflects the interaction between individual susceptibility and environmental factors.
1. Rate of acute lymphocytic leukemia
The American Cancer Society (ACS) estimates that in the US there were approximately 6020 new cases of ALL in 2014 (3140 men and 2880 women) and approximately 1440 deaths from ALL (810 men, 630 women). Most cases of ALL occur in children, but deaths from ALL are mainly in adults (4/5).
The age-adjusted incidence of ALL is 1.6/100,000 men/year and 1.2 for women in the US, based on cases diagnosed between 1975 and 2010. The risk of ALL is highest in children. under 5 years old. The risk then decreases slowly until about age 20, and increases slowly after age 50. Rates are 7.9/100,000 children 1 to 4 years old and 1.2 for adults over 60. Only 20% of acute leukemias in adults are ALL, but one-third of ALL cases are adults. The mean lifetime risk of ALL is <1/750. The risk is higher in men than in women, and in white versus black Americans.
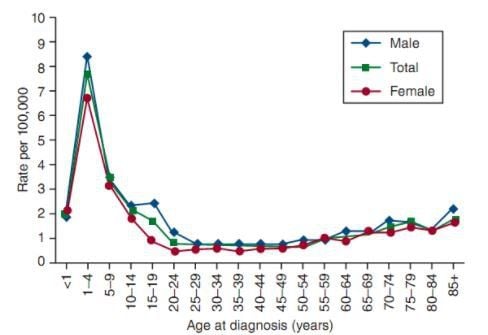
Hình 2: Tỷ lệ ALL theo từng độ tuổi và theo giới. (Dữ liệu từ SEER Cancer Statistics Review, 1975–2010, National Cancer Institute. Bethesda, MD, http://seer.cancer.gov/ csr/1975_2010. Accessed July 4, 2014)
The mean age at diagnosis of ALL is 13 years and approximately 61% are diagnosed before the age of 20. But because it has two peaks, the number 13 is only mathematically correct and has almost no medical significance. ALL is the most common malignancy diagnosed in young adults <15 years of age, with an estimated 23% of all cancers and 76% of leukemia in this age group.
Sharp peaks in the incidence of ALL in children have been observed in the United Kingdom and the United States since 1930. In the United States, the first peak appears to be observed in children of European ancestry, followed by the group of children. of African descent in the 1960s. The age-related peak is almost unobserved in many developing or undeveloped countries, suggesting that a contributing factor in the pathogenesis of leukemia is related to industrialization. With the exception of a slight predominance in females in the neonatal population, ALL affects males of European ancestry with a predominance in all age groups.
The incidence of ALL varies widely across geographic regions. Prevalence is high in residents of Northern Europe, Western Europe, North America, Oceania and lower in Asia and Africa.
2. Risk factors for acute lymphoblastic leukemia
2.1. Genetic syndromes The exact pathogenesis of ALL is poorly understood. Only a small percentage (5%) are linked to genetic syndromes. Children with Down syndrome are 10-30 times more likely to have leukemia; Acute platelet progenitor leukemia mainly in children <3 years of age and ALL mainly in the older age group. ALL in patients with Down syndrome is a heterogeneous disorder that includes subgroups with established genetic abnormalities similar to those found in the general population of >50 chromosomal polyploidy and t(12;21)[ ETV6-RUNX1] and common abnormalities associated with Down syndrome such as +X, del(9), and CEBPD rearrangements. Studies have shown that recombinant P2RY8-CRLF2 and activating JAK mutations contribute to leukemia in approximately half of all Down syndrome patients with ALL. Almost all patients with ALL with Down syndrome have a deletion of the IKZF1 gene. Autosomal recessive disorders that are often associated with increased chromosomal fracture susceptibility and predisposition to ALL are ataxia-telangiectasia (also known as Louis-Bar syndrome), Nijmegen rupture syndrome (Nijmegen breakage syndrome) and Bloom syndrome. Patients with ataxia-vasodilatation have a 70-fold increased risk of leukemia and a 250-fold increased risk of lymphoma, particularly the T-cell phenotype. The causative gene, ATM (ataxia-telangiectasia mutated), codes for the protein. involved in DNA repair, regulation of cell proliferation and apoptosis. Experimental studies that support the diagnosis of ataxia-vasodilatation include increased α-fetoprotein levels, presence of characteristic chromosomal abnormalities, absence or decreased serine protein kinase ATM in the nucleus, and increased sensitive to radiation in vitro. A high rate of ATM gene alterations seen in children with T-ALL suggests a pathophysiological role for ATM in lymphoid malignancies. Although immunodeficiency contributes to the increased risk of EBV-associated malignancies in acquired immunocompromised individuals, there is no convincing evidence that immunodeficiency plays a role in the increased risk of EBV infection. ALL in patients with ataxia-vasodilatation or other hereditary immunodeficiency syndromes. Genome-wide correlation study, the common allele variation in 4 genes (IKZF1, ARID5B, CEBPE, CDKN2a) has a constant association with ALL in children.
2.2. Environmental factors Exposure to X-rays in utero (but not after birth) is associated with a slightly increased risk of ALL, which is positively correlated with the number of exposures. Weak evidence for an association between ALL development and nuclear fallout; occupational, terrestrial or space-based ionizing radiation exposure; or exposure to paternal radiation before conception. There has been concern that exposure to low-energy electromagnetic fields generated by residential power supplies may be associated with the development of pediatric ALL. Case-control studies have shown a slightly increased risk of leukemia at very high exposures; Assuming the link is real, only about 1 percent of leukemia can be attributed to exposure. Pesticide exposure (occupational or home use) and parental smoking before or during pregnancy, infant vitamin K use, maternal alcohol consumption during pregnancy, and increased consumption dietary nitrites have been suggested. However, each of these links was controversial and most were disproved after careful, controlled investigation. High birth weight is associated with an increased risk of leukemia before age 5 with constant consistency, and birth weight may be indicative of an endogenous factor, such as insulin-like growth factor .
2.3. Host factors Genetic polymorphisms of xenobiotic-metabolizing enzymes, DNA repair pathways, and cell cycle checkpoint functions may interact with environmental, dietary, maternal and other factors Other external factors influence the development of ALL. Although the number of studies and sample size are limited, data exist that support a causal role for polymorphisms in genes encoding detoxifying enzymes (eg, glutathione S-transferase, nicotinamide adenine dinucleotide phosphate) [NAD(P)H]: quinone oxidoreductase), folate-metabolizing enzymes (serine hydroxymethyltransferase and thymidylate synthase), cytochrome P450, methylenetetrahydrofolate reductase, and cell cycle inhibitors in ALL development in children and adults. However, all of these associations must be confirmed by larger studies with careful attention to ethnic and geographic diversity in polymorphism frequency. Using genome-wide analysis, single nucleotide polymorphisms (SNPs) of AT-rich interactive domain 5b (ARID5B) were associated with polyploid B-cell precursor ALL in children, a clear example of variables Host genetics influence the development of ALL children.
2.4. Fetal ALL formation A retrospective review of leukemia-specific fusion genes (eg, KMT2A/AFF1 [also known as MLL-AF4] and ETV6-RUNX1 [also known as TEL-AML1]), polyploidy, or Ig/TCR rearrangement of stored neonatal blood drops (Guthrie cards) and the development of concomitant leukemia in identical twins clearly indicate some leukemia of prenatal origin. In identical twins with t(4;11)/KMT2A/AFF1, the concordance rate is close to 100 percent, and the latency in time to occur in the twins is short (several weeks to months). These findings suggest that this fusion gene alone is either leukemogenic or only requires a small number of mutations to cooperate to cause leukemia. In contrast, the lower concordance rates in twins with either the ETV6-RUNX1 fusion or the T-cell phenotype and a longer postpartum latency suggest that additional postnatal events are required for leukemic transformation in these subtypes. This theory is supported by the identification of rare cells expressing ETV6-RUNX1 fusion copies in approximately 1 percent of neonatal cord blood samples, a frequency 100-fold higher than the incidence of ALL identified. identified by this fusion copy.

Sự hình thành ALL trong bào thai là một trong các yếu tố nguy cơ bệnh bạch cầu cấp tính dòng Lympho
Presence of preleukemic clone with ETV6-RUNX1 has been established. Polyploid ALL, another common subtype of pediatric ALL, is also present before birth but requires postnatal events for complete malignant transformation. Observations on the peak of ALL development in children 2 to 5 years of age, association with an industrialized, modernized, or affluent society with increased rates of ALL, and sporadic cases of childhood leukemia have been reported. promotes two parallel infection-based hypotheses to account for postnatal events. The "delayed-infection" hypothesis proposes that some individuals susceptible to prenatally acquired preleukemic clones have little or no exposure to common infections early in life due to living in highly hygienic environments. Isolating such infections results in the immune systems of these individuals reacting abnormally or pathologically after repeated or late exposure to common infections at an age commensurate with lymphocyte proliferation. . The “population-mixing” hypothesis predicts that pediatric ALL is the result of exposure of susceptible (non-immune) individuals to common (but not immune) infectious agents. completely pathogenic) after mixing with healthy carriers. However, it is clear that not all cases of childhood leukemia develop in utero. For example, t(1;19)/TCF3-PBX1 (also known as E2A-PBX1) ALL appears to be of postnatal origin in most cases. Adult cases of ALL inevitably arise over an extended period of time.
3. Acquired Genetic Changes
Acquired genetic abnormality is a hallmark of ALL; 80 percent of all cases have recurrent cytogenetic or molecular lesions that are prognostic and treatment-related. Chromosomal changes include abnormalities in the number (ploidy) and structure of chromosomes. Structural abnormalities include translocation (most common), inversion, deletion, point mutation, and amplification. Although the frequencies of specific genetic subtypes vary between children and adults, the general mechanisms of effect are similar. Mechanisms include abnormal expression of oncoproteins, loss of tumor suppressor genes, and chromosomal translocations that induce fusion genes encoding transcription factors or active kinases.
Primary genetic rearrangement by itself is not sufficient to cause overt leukemia. Collaborative mutations are required for leukemia transformation and include genetic and epigenetic changes in important growth regulatory pathways. The candidate gene approach clearly identified the deletion of CDKN2A/CDKN2B tumor suppressor loci and NOTCH1 mutations in T-cell ALL. Current searches using genome-wide microarray and high-throughput sequencing have identified a high frequency of genetic alterations common to both B-cell precursor ALL and T-cell ALL. Using the SNP microarray, an average of 6.46 DNA copy number abnormalities (CNAs) for each case were identified, suggesting that overall genomic instability is not a feature for most cases. There is a large variation in the number of CNAs between the leukemic subtypes. Interestingly, the neonatal ALL cases with MLL rearrangement had less than one CNA per case, suggesting that less additional genetic damage was needed to develop leukemia in this case. In contrast, cases ETV6-RUNX1 and BCR-ABL1 had more than six CNAs each, with some cases having more than 20 lesions, a finding consistent with that notion. Although the initiating events may occur early in childhood, additional subsequent lesions are required to develop ALL. More than 40% of cases of B-cell precursor ALL have mutations in genes that encode substances that regulate normal lymphoid growth. The most common target is the lymphoid transcription factor PAX5 (mutated in about 32% of cases), which encodes the paired-domain protein required for pro-B-cell to pre-B-cell conversion and B-lineage stabilization. The second most frequently implicated gene is IKZF1 (mutated in nearly 28% of cases), which encodes the IKAROS zinc finger DNA-binding protein required for earliest lymphoid differentiation. IKZF1 was deleted in the majority of cases of BCR-ABL1 ALL as well as in the lymphoid blast crisis phase of chronic myeloid leukemia (but not in the chronic phase). About half of BCR-ABL1 ALL cases also have CDKN2A/B and PAX5 deletes. This finding further supports the idea that multiple signaling pathways need to be disrupted to cause leukemia. A subgroup of ALL with very poor results was strongly associated with the presence of delete IKZF1. All these findings suggest that IKZF1 is directly related to treatment resistance in ALL.
BCR-ABL1-like B-cell ALL fusion BCR-ABL1 or t (9; 22) by cytogenetic, FISH or molecular analysis, but it shares the same gene-expression profile with typical BCR-ABL1–positive ALL. In half of these cases, the CRLF2 gene was associated with a hidden translocation with the IGH gene or was fused with the P2RY8 gene; both rearrangements resulted in CRLF2 overexpression. Mutations in JAK2 or JAK1 were detected in 30 to 40 percent of these cases, and many of the remaining cases triggered mutations in the cytokine receptor and kinase signaling pathways. Microarrays and genomic DNA sequencing identified monoallelic deletion of the PAX5 gene at 9p13.2 in 28 percent of ALL patients with hidden deletions or larger fragments in 9p.
Gene-expression profiling with DNA microarrays allows nearly all cases of T cells to cluster along multistage oncogenic pathways. Gene expression studies also revealed overexpression of FLT3, a receptor tyrosine kinase important for the development of hematopoietic stem cells, which is a secondary event in most cases with the development of hematopoietic stem cells. MLL or hyperdiploidy rearrangement. This finding provided an impetus for clinical trials of FLT3 inhibitors in ALL. Genome-wide interactions of both leukemia cells and germline tissues have identified other genetic variants that are relevant for prognosis or treatment and may lead to the development of specific therapies.
Epigenetic changes, including hypermethylation and silencing of tumor suppressor genes and hypomethylation of oncogenes and abnormalities in post-transcriptional control mechanisms, such as those involved in microRNAs, are common findings variable in cancer. These changes are reversible and do not alter the DNA sequence, but they can alter gene expression in subtle ways that encourage mutation and malignant progression. The analysis of epigenetic changes has begun to be applied to develop novel biomarkers for risk stratification or disease monitoring and to design alternative treatment in ALL. Evidence indicates that methylation of multiple genes in ALL is associated with a worse outcome. Surprisingly, gene methylation was as prominent in ALL children as adults. Differences in the responses of children and adults do not appear to be related to total methylation but to specific genes and specific pathways being disabled. Preliminary studies of hypomethylating agents (eg, azacitidine and decitabine) are being tested in patients resistant to current agents.

Please dial HOTLINE for more information or register for an appointment HERE. Download MyVinmec app to make appointments faster and to manage your bookings easily.