This is an automatically translated article.
Posted by Master, Doctor Vu Duy Dung - Department of General Internal Medicine - Vinmec Times City International HospitalAtaxia is the term for a group of disorders that affect coordination, balance, and voice. Symptoms and severity of ataxia depend on the cause of the disease, including genetic ataxia and degenerative ataxia.
1. Genetic ataxia
There are many genetic causes of ataxia. The Human Online Mendelian Genetics Data (OMIM) can be a useful and up-to-date source of information for ataxia genes. What follows is an overview of ataxia genes based on genotype.
1.1 Autosomal dominant ataxia With autosomal dominant ataxia, the term SCA has been named, from types 1 to 48 to this day. Of these, cerebellar ataxia caused by prolongation of CAG repeats, including SCA1, SCA2, SCA3, SCA6, SCA7, and SCA17, is the most common, accounting for about half of all autosomal dominant ataxia. Other repeat-associated ataxia disorders often have repeat prolongation in noncoding regions of the gene, such as SCA8, SCA10, and SCA12. The relatively rare remainder are SCAs that are often associated with sequence variations in the coding region. Note that whole-exon sequencing is best for detecting gene sequence variations but not repeat elongation. Therefore, identifying repeat prolongation in common SCA genes is the first step in diagnosing SCA genes. If no repeat stretch is found, sequencing methods should be the next step.
Each SCA has distinct but also overlapping clinical features. Early SCA1 patients have dysphagia and difficulty speaking. Patients with SCA2 often have slow gaze, grogginess, hyporeflexia, and tremors. Patients with SCA3 often have dystonia, depression, restless legs syndrome, and Parkinson's syndrome. Patients with classic SCA6 have only cerebellar ataxia without other extracerebellar symptoms. Patients with SCA7 have vision loss due to retinitis pigmentosa. Patient SCA17 has dementia, chorea, and dystonia. Although these clinical features are useful in guiding the correct genetic diagnosis, there is a strong racial preference. For example, SCA2 is very common in Cuba, whereas SCAC3 is most common in China, Portugal, and Brazil. Different types of SCAs with CAG repeats have different rates of disease progression. For example, SCA1 progressed fastest, followed by SCA3 and SCA2. SCA6 has the slowest progression of these types.

Vị trí các đoạn lặp CAG trong bệnh thất điều tiểu não
1.2 Autosomal recessive ataxia can usually be divided into three groups:
(1) Cerebellar ataxia with predominance of sensory neuropathy (2) Cerebellar ataxia with Sensory axonal neuropathy (3) Cerebellar ataxia without sensory neuropathy. Therefore, the features of the associated neuropathy are key to examining recessive ataxia. The first disease for the cerebellar ataxia group with a predominance of sensory neuropathy was Friedreich's ataxia, the most common recessive ataxia. Patients with Friedreich's ataxia have an age of onset from infancy to the third decade and may also have concave feet, scoliosis, square wave nystagmus, and decreased tendon reflexes. Approximately 15% of cases of Friedreich's ataxia are adult-onset, which may be accompanied by spasticity and markedly increased tendon reflexes. Diabetes and cardiomyopathy are also common in Friedreich's ataxia. Friedreich's ataxia is caused by elongation of non-coding GAA repeats of the FXN gene, which results in insufficient frataxin production and leads to mitochondrial dysfunction. Approximately 5% of patients with Friedreich's ataxia have one allele carrying a point mutation in the FXN gene and the other allele with repeat prolongation. Therefore, a neurologist should still have a high suspicion of Friedreich's ataxia if duplication prolongation is detected in only one allele in a patient with classic clinical symptoms of Friedreich's ataxia.

Bệnh nhân thất điều Friedreich có thể có biểu hiện vẹo cột sống
Another relatively common recessive ataxia is SANDO syndrome, caused by mutations in the POLG gene. Age of onset is adult with ophthalmoplegia, ptosis, myoclonus, and epilepsy. Hyperintensity on T2 of the bilateral inferior squamous nuclei can sometimes be seen on brain MRI.
Another group of autosomal recessive ataxia is cerebellar ataxia with motor-sensory axonal disease. In this group, the most common forms of the disease are stellate ataxia and ataxia with loss of ophthalmomotor use type 1 and type 2. In addition to neuropathy, chorea, dystonia, and spasticity are the symptoms are common in this group. Levels of α-Fetoprotein are increased in stellate ataxia and ataxia with type 2 oculomotor ataxia, which can sometimes be used as a screening tool for these diseases. Patients with atrial fibrillation have an increased risk of cancer and infection, along with radiation sensitization.
The third group of recessive ataxia is ataxia without sensory neuropathy, in which autosomal recessive cerebellar ataxia type 1 is usually adult-onset with upper or lower motor neuron signs. , concave feet, and scoliosis. Patients with Niemann-Pick disease type C have characteristic supranuclear longitudinal ophthalmoplegia along with mental retardation, dystonia, and cognitive impairment.
1.3 X-linked ataxia The most common X-linked ataxia is fragile X syndrome (fragile X) ataxia, caused by elongation of the CGG repeat in the FMR1 gene , leading to toxic RNA function. A further repeat prolongation of the FMR1 gene would, as expected, lead to premature ovarian failure in the patient's daughters and mental retardation in the patient's grandsons.
Therefore, fragile X syndrome should be placed in the context of a family history of mental retardation and premature ovarian failure. Parkinson's syndrome and autonomic dysfunction may also be present; Therefore, the fragile X tremor-ataxia syndrome should be included in the differential diagnosis of multiple systems atrophy. T2 hyperintensity in the midcerebellar peduncles and corpus callosum on brain MRI are characteristic of the fragile X-chromosome tremor syndrome and may aid in the diagnosis.
1.4 Mitochondrial ataxia Mutations in mitochondrial DNA can also cause ataxia. Common causes are Kearns-Sayre syndrome, myoclonic epilepsy with tattered red fibers (MERRF), and mitochondrial encephalopathy, lactic acidosis, and stroke-like episodes (MELAS). Because of tissue-specific mitochondrial defects (that is, cytoplasmic heterogeneity), muscle biopsies should be performed because mitochondrial defects are more likely to be detected in nondividing cells, such as muscle cells.
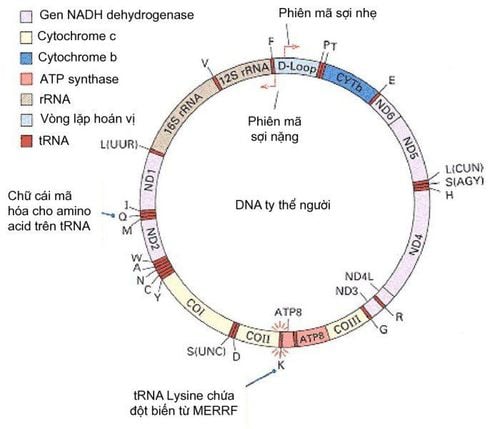
Bản đồ Gene của DNA ty thể trong cơ thể con người
1.5 Episodic ataxia Episodic ataxia (types 1 to 8) constitutes a group of genetic causes of episodic ataxia, lasting a few minutes to several days. There may be progressive background ataxia with exacerbations. Some episodes of ataxia can be triggered by exertion. Gene mutations associated with ataxia are often in ion channels or membrane proteins important for neuronal excitability. The main differential diagnoses considered for episodic ataxia are multiple sclerosis and radiculopathy.
1.6 A Rational Approach to Diagnosing Genetic Ataxia Despite rapid advances in genome-wide and exon sequencing technologies, the first approach to diagnosing genetic ataxia is a genetic test for ataxia. Duplicate lengthening because ataxia associated with this repeat is the most common (autosomal dominant: SCA1, SCA2, SCA3, SCA6; autosomal recessive gene: Friedreich ataxia; chromosomal linkage ataxia: X body: fragile X syndrome ataxia), and changes in repeat length would not be detected by sequencing. After excluding repeat-associated ataxias, whole-exon sequencing can be very useful for identifying gene sequence variations. Another limitation of whole-exon sequencing for ataxia is that this method is not sensitive to large chromosomal deletions or duplications, as in SCA20, or to ataxia associated with mitochondrial gene mutations. , or with loop stretching in non-coding regions. A diagnostic test for ataxia should be accompanied by clinical presentation for appropriate genetic testing.
Researchers performed genetic sequencing analysis in a large cohort study of ataxia patients with no family history. It turns out that autosomal recessive type I cerebellar ataxia with mutations in the SYNE1 gene is the most common and, therefore, should always be considered when experiencing adult-onset ataxia with polymotor neuron signs. patterns such as increased tendon reflexes, extensor foot responses, muscle atrophy, or fasciculations. It has also recently been recognized that genetic mutations in spastic biplegia sometimes cause cerebellar ataxia. While these gene mutations follow the path of not being grouped in SCAs or recessive ataxias, physicians must always keep these genes in mind. An example is the SPG7 mutation, which has been seen in patients with idiopathic cerebellar ataxia.

Sơ đồ các đột biến được tìm thấy trong nghiên cứu liên quan đến các miền SYNE1
2. Ataxia due to degeneration
Degenerative forms of ataxia are common in patients over 60 years of age with no family history of ataxia. In this group, multiple system atrophy and late-onset idiopathic cerebellar ataxia are the two most common diseases.
Multisystem atrophy characterized by cerebellar ataxia, Parkinson's syndrome, autonomic dysfunction (postural hypotension, impotence, and polyuria, urgency, and incontinence), and signs tower sign. Depending on which symptom is more prominent, multisystem atrophy can be subdivided into Parkinson's-type multisystem atrophy or cerebellar-type multisystem atrophy. The cerebellar type multisystem atrophy accounts for about 30% of all cases of multiple system atrophy. Patients with multiple-system atrophy with predominant cerebellar ataxia generally have a slower progression than patients with dominant parkinsonism. In the early stages of the disease, patients may have relatively isolated cerebellar ataxia.
Therefore, the history and examination should focus on looking for any signs of Parkinson's syndrome, autonomic dysfunction, and upper motor neuron signs. REM sleep behavior disturbances are also common, which can be useful to indicate an underlying synuclein disease associated with multiple system atrophy. Stridor and sleep apnea are sometimes present. Therefore, in patients with late-onset cerebellar ataxia, autonomic and sleep investigations may provide additional evidence to support the diagnosis of multisystem atrophy when clinical symptoms are unclear. Brain MRI in patients with multiple system atrophy may present with hot crosshairs; This sign may not be obvious in the early stages of the disease but may become apparent later in the disease.
Multiple system atrophy is a relatively rapidly progressive disease. However, late-onset idiopathic cerebellar ataxia is a slowly progressive disease, and it usually does not reduce life expectancy. Patients with late-onset idiopathic cerebellar ataxia usually will not have other neurological signs such as tendon hyperreflexia or Parkinson's syndrome.

Một số triệu chứng thường gặp ở người bệnh mắc hội chứng Parkinson
Neurological examination confirmed ataxia. To register for examination and treatment at Vinmec International General Hospital, you can contact the nationwide Vinmec Health System Hotline, or register online HERE.
Source: Kuo SH. Ataxia. Continuum 2019;25(4, Movement Disorders): 1036–1054.
See more Documentation about Ataxia Master Vu Duy Dung:
Symptoms and causes of ataxia Clinical examination, laboratory tests and diagnosis of ataxia Genetic ataxia and degenerative ataxia ataxia treatment