Bài viết bởi Tiến sĩ Hàn Thị Thu Hương - Khối Di truyền y học, Trung tâm Công nghệ cao Vinmec.
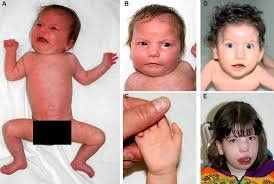
Nhiễm sắc thể 18, Monosomy 18p hay còn được gọi là Hội chứng mất đoạn 18p là một rối loạn nhiễm sắc thể hiếm gặp do bị mất một phần hoặc cả nhánh ngắn nhiễm sắc thể 18. Hội chứng này thường được đặc trưng bởi tầm vóc thấp bé, chậm phát triển trí tuệ, chậm nói, dị dạng vùng sọ, vùng mặt và các bất thường về thể chất khác.
1. Nhiễm sắc thể 18, Monosomy 18p
Monosomy 18p xuất hiện với tỉ lệ khoảng 1:50.000 trẻ sinh sống với tỉ lệ nữ và nam tương ứng khoảng 3:2. Kể từ khi hội chứng này được mô tả ban đầu vào năm 1963, đến nay đã có hơn 150 trường hợp được báo cáo trong các tài liệu y tế.
2. Dấu hiệu và triệu chứng
Trong nhiều trường hợp, trẻ sơ sinh mắc Monosomy 18p có trọng lượng sơ sinh thấp; thiếu hụt tăng trưởng từ nhẹ đến trung bình; giảm trương lực cơ; chậm phát triển trí tuệ mức độ trung bình; chậm tiếp thu các kỹ năng nói và ngôn ngữ, không nói được những từ hoặc câu đơn giản trước khoảng bảy đến chín tuổi. Một số trẻ có thể có những bất thường về hành vi hoặc cảm xúc như khó tập trung, bồn chồn và tâm trạng thay đổi nhanh chóng (cảm xúc không ổn định).

Mức độ bất thường sọ não thay đổi tùy trường hợp bao gồm: đầu nhỏ bất thường (microcephaly); khuôn mặt tròn đặc biệt; miệng rộng, “hình cá chép”; mũi tẹt hoặc rộng; tai lớn, dị dạng, nằm thấp hơn bình thường; hai mắt cách xa nhau (ocular hypertelorism), sụp mí mắt trên (ptosis), mắt lác; hàm dưới nhỏ bất thường, thụt vào trong (microretrognathia).
Khoảng 10% người mang mất đoạn 18p mắc “holoprosencephaly”, một bất thường bẩm sinh phức tạp của não do não trước không thể phân chia bình thường thành các bán cầu trong quá trình phát triển phôi thai. Holoprosencephaly có thể dẫn đến chậm phát triển trí tuệ ở các mức độ khác nhau, động kinh và các triệu chứng thần kinh khác. Holoprosencephaly cũng có thể dẫn đến các bất thường trên mặt như khuyết tật đường giữa khuôn mặt từ nhẹ đến nặng, chỉ có một răng cửa duy nhất ở hàm trên (maxillary incisor); mắt cách xa hoặc quá gần nhau (ocular hypertelorism or hypotelorism); trong những trường hợp cực kỳ nghiêm trọng, các hốc mắt hợp nhất thành một hốc duy nhất chứa một mắt (cyclopia); hở hàm ếch; mũi tẹt bất thường; có một lỗ mũi hoặc không có mũi (arhinia).
Một số trẻ sơ sinh có thể bị tích tụ chất lỏng bất thường trong các mô mềm của bàn tay và bàn chân, kèm theo sưng tấy (phù bạch huyết); cổ ngắn, có màng (webbed neck); ngực rộng bất thường, bàn tay và bàn chân tương đối nhỏ; ngón tay ngắn; quẹo "ngón út" (clinodactyly). Nam giới mắc bệnh có thể có dương vật nhỏ bất thường (micropenis), tinh hoàn ẩn (cryptorchidism). Một số trường hợp thiếu hụt kháng thể (immunoglobulin A); dị tật tim bẩm sinh.
3. Nguyên nhân

Hầu hết mọi người được sinh ra với 23 cặp (tổng cộng 46) nhiễm sắc thể trong mọi tế bào của cơ thể. Mỗi nhiễm sắc thể bao gồm một nhánh dài (q) và một nhánh ngắn (p) được ngăn cách với nhau bởi tâm động (centromere). Monosomy 18p là bệnh lý nhiễm sắc thể do mất cả hoặc một phần nhánh ngắn trên nhiễm sắc thể 18. Rối loạn này thường không phải do di truyền, mà xảy ra do sự phát sinh ngẫu nhiên (de novo) không rõ nguyên nhân rất sớm trong quá trình phát triển phôi thai. Trong những trường hợp như vậy, cha mẹ của đứa trẻ bị đột biến thường có nhiễm sắc thể bình thường và nguy cơ sinh con khác bị bất thường nhiễm sắc thể tương đối thấp.
Trong một số trường hợp mất đoạn 18p dường như là kết quả của đột biến “chuyển đoạn cân bằng” trong các cặp bố mẹ. Nếu sự sắp xếp lại nhiễm sắc thể là cân bằng, nghĩa là bộ nhiễm sắc thể bị thay đổi nhưng không thừa hoặc thiếu vật chất di truyền thì nó có thể vô hại đối với người mang đột biến tuy nhiên có thể tăng nguy cơ phát sinh nhiễm sắc thể bất thường ở người con.
Ngoài ra, trong một số trường hợp hiếm hoi, cha mẹ của một đứa trẻ bị bệnh cũng mang mất đoạn 18p ở tất cả hoặc một số tế bào (dạng khảm).
4. Chẩn đoán
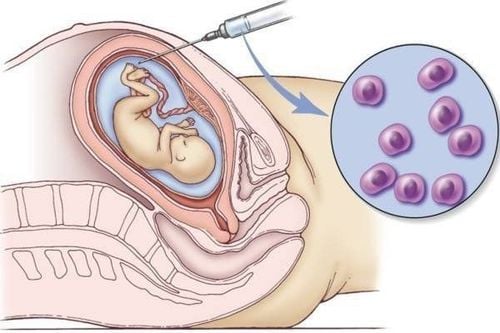
Hội chứng Monosomy 18p có thể được chẩn đoán trước sinh bằng siêu âm, chọc dò màng ối và sinh thiết gai nhau (CVS) hoặc sau sinh trên mẫu máu ngoại vi để thực hiện các xét nghiệm di truyền tế bào như Karyotyping (Công thức nhiễm sắc thể), FISH (Lai huỳnh quang tại chỗ), Array-CGH (Lai so sánh hệ gen).
5. Điều trị
Vì Hội chứng Monosomy 18p ảnh hưởng rất khác nhau trên mỗi cá thể nên việc điều trị và can thiệp phải được điều chỉnh theo nhu cầu từng cá nhân mắc bệnh. Việc điều trị có thể đòi hỏi sự phối hợp của một nhóm chuyên gia y tế, như bác sĩ nhi; bác sĩ phẫu thuật; bác sĩ chuyên về các rối loạn của khung xương, cơ, khớp và các mô liên quan (bác sĩ chỉnh hình); nhà thần kinh học; nhà bệnh lý học ngôn ngữ - lời nói và các chuyên gia chăm sóc sức khỏe khác.
Sự can thiệp sớm rất quan trọng trong việc giúp trẻ em mắc hội chứng mất đoạn 18p có thể phát triển bình thường. Phương pháp chủ yếu là giáo dục đặc biệt, trị liệu ngôn ngữ, vật lý trị liệu, các dịch vụ y tế, xã hội và dạy nghề khác. Tư vấn di truyền được khuyến nghị cho gia đình có trẻ mang Hội chứng Monosomy 18p.
Để đặt lịch khám tại viện, Quý khách vui lòng bấm số HOTLINE hoặc đặt lịch trực tiếp TẠI ĐÂY. Tải và đặt lịch khám tự động trên ứng dụng MyVinmec để quản lý, theo dõi lịch và đặt hẹn mọi lúc mọi nơi ngay trên ứng dụng.
Tài liệu tham khảo: rarediseases.org