This is an automatically translated article.
Post by Master Doctor, Doctor Mai Vien Phuong - Head of Department of Gastrointestinal Endoscopy - Department of Medical Examination & Internal Medicine - Vinmec Central Park International General Hospital
Amyloidosis is defined as extracellular deposition of unbranched fibers with a β-pleated plate configuration. It can be acquired or inherited and systemic or localized. The common forms of systemic amyloidosis are: primary, secondary, dialysis-associated, and familial polyneuropathy.
1. Overview of gastrointestinal amyloidosis
Primary amyloidosis involves monoclonal light chains in serum or urine in 15% of patients with multiple myeloma. Secondary amyloidosis is caused by the deposition of amyloid A (AA) protein, which is the result of proteolytic cleavage of circulating acute-phase reactive serum amyloid A. Secondary amyloidosis is associated with a variety of disorders. chronic inflammatory disorders, chronic bacterial infections or, less commonly, malignancies. The clinical presentation of amyloidosis can vary from asymptomatic to fatal. Amyloidosis is one of those unusual diseases that doctors may not think about when it affects patients. Advances have been made in the understanding of the pathogenesis, clinical features, diagnostic measures, and therapeutics of this disorder. Here, we will discuss the clinical management of gastrointestinal (gastrointestinal) amyloidosis.
Amyloidosis is a rare disease, the exact incidence of which is unknown. Most studies show that the disease is not related to race, occupation, or environmental factors. Some statistics show that the disease is more common in men, less common under 40 years old. Among the systemic forms, the AL body (light chain Ig body) is the most common, accounting for about 60%. In the United States, there are an estimated 1500 to 2500 new cases of AL-type systemic amyloidosis each year, of which one-fifth is multiple myeloma. Localized amuliodosis accounts for less than 10% of all diagnosed patients. These genetic variants are quite rare and are mostly autosomal dominant.

Một số thống kê cho thấy bệnh amyloidosis đường tiêu hóa gặp nhiều hơn ở nam giới, ít gặp dưới 40 tuổi
2. Clinical manifestations of Amyloidosis
The presentation of gastrointestinal amyloidosis can depend on the amount and location of amyloid deposition regardless of whether it is primary or secondary amyloidosis, and varies from asymptomatic to fatal.
2.1 Systemic manifestations Systemic amyloidosis with gastrointestinal involvement predominates in 60 (79%) patients. Of the 60 systemic cases, 50 (83%) had immunoglobulin light chain disease, 5 (8%) familial lysozyme, 5 (8%) transthyretin amyloidosis. The most common symptoms for all patients were weight loss in 33 (45%) and gastrointestinal bleeding in 27 (36%). Random identification of amyloidosis on routine endoscopic surveillance played a role in the diagnosis of 11 patients. Although a rare manifestation of amyloidosis, amyloid staining should be considered in patients undergoing gastrointestinal biopsy with unexplained chronic gastrointestinal symptoms.
2.2 Kidney damage Umbrella is the most common finding present in all types of amyloidosis. Involvement of the kidney, heart, and peripheral nerves leads to the general presentation of symptoms. Renal damage is initially characterized by proteinuria, with uremia developing late in the course of the disease.
2.3 Cardiac damage Cardiac deposition leads to restrictive cardiomyopathy with congestive heart failure and subsequent various arrhythmias.
2.4 Lesions of the mouth The most common oral manifestation of amyloidosis is macroglossia, which can cause sleep apnea, difficulty speaking, difficulty swallowing in the mouth, difficulty chewing, mismatched teeth, indented teeth, and finally, airway obstruction.
2.5 Skin lesions Papules, vesicles and sores can be seen. There may be swelling of the floor of the mouth, stiffness of the soft tissues around the mouth, loss of facial expression, and difficulty opening the mouth. The subchondral glands may be involved, leading to xerostomia, like Sjogren's syndrome.
2.6 Gastrointestinal lesions Patients with gastrointestinal amyloidosis may present with abdominal pain, dysfunction, diarrhea, bleeding, pseudo-obstruction, and malabsorption. The most common presenting symptoms are fatigue, lightheadedness, and weight loss.
Amyloidosis of the gastrointestinal tract, with biopsy-proven disease, is rare. In the latest gastrointestinal amyloidosis study, out of 2334 patients with all types of amyloidosis, 76 patients (3.2%) had biopsied involvement of amyloid in the gastrointestinal tract. chemical. The mean age is 61 years old.

Bệnh nhân mắc bệnh amyloidosis đường tiêu hóa có thể có biểu hiện đau bụng, rối loạn chức năng, tiêu chảy, chảy máu, giả tắc nghẽn và kém hấp thu
2.7 Damage to the esophagus Esophageal disease in amyloidosis includes symptoms such as dysphagia, chest pain, heartburn, and regurgitation. The most common radiographic feature is dilated esophagus, atony with decreased motility, sometimes with distal stenosis and proximal dilatation, associated with bronchoscopy. Less common findings are sores or masses. A variety of pressure abnormalities can be seen in the esophagus. Generally, lower esophageal sphincter pressure is normal or low, often accompanied by heartburn. The amplitude of contractions is reduced. Occasionally, the images may mirror those seen in achalasia. Unlike idiopathic achalasia, patients have a rapid onset of symptoms and weight loss. Nerve involvement in esophageal amyloidosis is thought to be due to the lack of expected secondary peristalsis and LES dilation induced by esophageal distension. Rates of esophageal disease in amyloidosis ranged from 13% in a radiological study to 22% in an autopsy series.
2.8 Stomach injury In patients with amyloidosis, gastric adhesions occurred in 8% of patients at biopsy and 12% at autopsy, with only 1% being symptomatic. Stomach-related symptoms include nausea, vomiting, regurgitation, and epigastric pain. Gastric outlet obstruction, gastric folds, gastric ulceration, hematoma, arteriovenous malformations, granular mucosa, plaque lesions, and gastroparesis may occur rarely.
2.9 Injury to the small intestine The greatest extent of amyloid deposition is in the small intestine, with 31% of patients with amyloid affected at autopsy. The vascular system frequently involved is usually located in the submucosa. The patient had amyloidosis of the small intestine with diarrhea, hypersecretion, protein loss, hemorrhage, obstruction, infarction, perforation, intussusception, pneumonia, and constipation. Poor absorption has also been found in some patients with amyloidosis.
2.10 Colon injury Clinical manifestations of colonic amyloid deposition may resemble other diseases including inflammatory bowel disease, malignancy, ischemic colitis, and spastic colitis . Complications include dilated colon, pseudo-obstruction, lumbar formation, rectal bleeding, submucosal hemorrhage, myocardial infarction, infarction, and perforation.
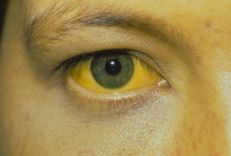
2.11 Liver damage Liver amyloidosis is of no clinical significance in the majority of patients, although symptomatic patients may present with weight loss, fatigue, abdominal discomfort, and anorexia (26%). Familial amyloidotic polyneuropathy affecting mainly the liver; Other organs involved are the spleen, heart, thyroid, adrenal glands, and vitreous of the eye. Clinically, splenomegaly may be seen in patients with amyloidosis. Gallbladder involvement in amyloidosis is rare only in rare cases.
Amyloidosis can present a wide range of gastrointestinal disease symptoms with investigative studies showing a wide range of abnormalities. One should obtain immunofixed serum or urine samples as well as a biopsy of the gastrointestinal mucosa stained with Congo Red. While most gastrointestinal complications are controlled with symptoms, treatment depends on the type of amyloidosis. Causal therapy is reserved for a select few from the various subtypes of this disorder.
Please dial HOTLINE for more information or register for an appointment HERE. Download MyVinmec app to make appointments faster and to manage your bookings easily.
References
Hajime Isomoto*, Yasuhiro Kamo, Chun Chuan Chen, Kazuhiko Nakao. Clinical management of gastrointestinal amyloidosis. Open Journal of Gastroenterology. Vol.2 No.4(2012), Article ID:25047.8
Stermark, P. (2005) Aspects of human amyloids and their filamentous polypeptides. FEBS Journal, 272, 5942- 5949. doi: 10.1111/j.1742-4658.2005.05024.x
Glenner, GG (1980) Amyloid deposition and amyloidosis. The β-fibrilloses. New England Journal of Medicine, 302, 1283-1292. doi: 10.1056 / NEJM198006053022305
Kyle, RA and Gertz, MA (1995) Primary systemic amyloidosis: Clinical and laboratory features in 474 cases. Seminar on Hematology, 32, 45-59.