Bài viết được tư vấn chuyên môn bởi Bác sĩ chuyên khoa I Nguyễn Thị Mỹ Linh - Bác sĩ Nhi sơ sinh - Khoa Nhi - Sơ sinh - Bệnh viện Đa khoa Quốc tế Vinmec Đà Nẵng.
Hội chứng Smith-Magenis (SMS) là một rối loạn phát triển phức tạp, dẫn đến tình trạng nhiều hệ thống cơ quan trong cơ thể bị ảnh hưởng, được đặc trưng bởi một mô hình các bất thường ngay khi mới sinh (bẩm sinh) cũng như các vấn đề về hành vi và nhận thức.
1. Hội chứng Smith - Magenis là gì?
Hội chứng Smith-Magenis là một khuyết tật bẩm sinh do giảm tốc độ vi lượng hoặc bất thường của nhiễm sắc thể số 17. Các đặc điểm chính của Hội chứng Smith-Magenis (SMS) bao gồm khuyết tật trí tuệ nhẹ đến trung bình, chậm nói và kỹ năng ngôn ngữ, các đặc điểm trên khuôn mặt, rối loạn giấc ngủ và vấn đề về hành vi. Hội chứng Smith-Magenis ảnh hưởng đến ít nhất 1 người trên 25.000 người trên toàn thế giới.
Hội chứng Smith-Magenis là một tình trạng nhiễm sắc thể liên quan đến nhiễm sắc thể 17. Hầu hết những người bị SMS đều bị xóa bỏ vật liệu di truyền từ một vùng cụ thể của nhiễm sắc thể 17 (17p11.2). Mặc dù khu vực này chứa nhiều gen, nhưng gần đây các nhà nghiên cứu đã phát hiện ra rằng sự mất mát của một gen cụ thể mà axit retinoic 1 hoặc RAI1 gây ra là nguyên nhân gây ra hầu hết các biểu hiện đặc trưng của tình trạng này. Ngoài ra, các gen khác trong nhiễm sắc thể 17 góp phần vào sự biến đổi và mức độ nghiêm trọng của các đặc điểm lâm sàng. Việc mất các gen khác trong vùng bị xóa có thể giúp giải thích tại sao các đặc điểm của hội chứng Smith-Magenis lại khác nhau giữa các cá nhân bị ảnh hưởng. Một tỷ lệ nhỏ những người mắc hội chứng Smith-Magenis có đột biến gen RAI1 thay vì mất đoạn nhiễm sắc thể.
Những sự mất đoạn và đột biến này dẫn đến việc tạo ra một phiên bản bất thường hoặc không có chức năng của protein RAI1. RAI1 là một yếu tố phiên mã liên quan đến các thông điệp liên lạc giữa DNA và RNA.

SMS không có tính di truyền. Tình trạng này thường là kết quả của sự thay đổi di truyền xảy ra trong quá trình hình thành các tế bào sinh sản (trứng hoặc tinh trùng) hoặc trong quá trình phát triển sớm của bào thai. Những người mắc hội chứng Smith-Magenis thường không có tiền sử về tình trạng này trong gia đình họ.
2. Dấu hiệu nhận biết hội chứng Smith- Magenis
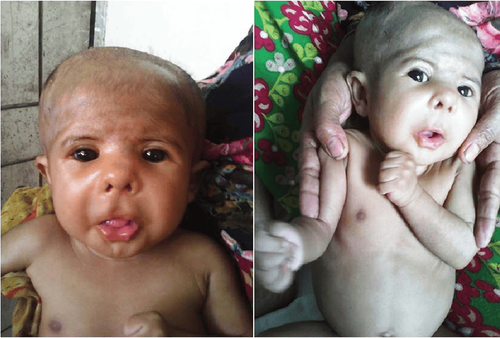
Những người mắc Hội chứng Smith-Magenis có đặc điểm là đầu phẳng ngắn, trán nổi rõ, khuôn mặt vuông rộng, mắt sâu, khuôn mặt kém phát triển, sống mũi rộng, mũi ngắn, môi trên hếch và cằm nhỏ từ khi còn nhỏ và trở nên tương đối nổi bật hơn so với lứa tuổi. Những khác biệt trên khuôn mặt này có thể là ít biểu hiện rõ khi trẻ ở thời thơ ấu, nhưng chúng thường trở nên đặc biệt hơn khi trẻ trưởng thành. Các bất thường về răng cũng thường xảy ra.
Trẻ mắc hội chứng Smith-Magenis trong thời kỳ sơ sinh thường có các vấn đề về bú, không phát triển như các bạn bè cùng lứa tuổi, trương lực cơ yếu, ngủ trưa kéo dài. Sau đó khi bước vào độ tuổi trưởng thành, giấc ngủ của trẻ sẽ bị rối loạn (khó đi vào giấc ngủ, thức giấc thường xuyên suốt đêm và buồn ngủ vào ban ngày), cũng như các vấn đề về hành vi.
Những vấn đề về hành vi này có thể được biểu hiện theo nhiều cách khác nhau, chẳng hạn như trẻ thường xuyên cáu giận, bốc đồng, lo lắng, mất tập trung, hung hăng và các hành vi tự gây thương tích bao gồm tự đánh, tự cắn và tự cấu.
Ngoài các hành vi thách thức nêu trên, trẻ mắc hội chứng Smith-Magenis còn có thể thể xuất hiện các hành vi khuôn mẫu bao gồm hành vi ngậm tay, ngậm đồ vật, đưa tay vào miệng, nghiến răng, đung đưa cơ thể và xoay tròn hoặc vật thể quay.
Các dấu hiệu và triệu chứng khác của hội chứng Smith-Magenis bao gồm tầm vóc thấp, độ cong bất thường của cột sống (vẹo cột sống), giảm độ nhạy cảm với cơn đau và nhiệt độ, có thể đi vệ sinh khó khăn và giọng nói khàn. Một số người bệnh cũng xuất hiện các bất thường về tai dẫn đến mất thính giác và có thể gặp khó khăn về thị lực. Mặc dù ít phổ biến hơn, tuy nhiên các khuyết tật về tim và thận cũng đã được báo cáo ở những người mắc hội chứng Smith-Magenis.

3. Chẩn đoán và điều trị hội chứng Smith-Magenis
Việc chẩn đoán Hội chứng Smith-Magenis (SMS) thường được thực hiện thông qua xét nghiệm máu lâm sàng được gọi là phân tích nhiễm sắc thể. Chẩn đoán cũng có thể được thực hiện thông qua xét nghiệm di truyền tế bào và FISH (lai huỳnh quang tại chỗ) hoặc bằng phân tích vi mảng nhiễm sắc thể (CGH).
Hầu hết những người được chẩn đoán mắc chứng SMS đều được sinh ra với sự xóa bỏ một phần nhỏ của cặp nhiễm sắc thể thứ 17 trong cơ thể. Chính việc thiếu hụt này, được gọi là 17p11.2, là nguyên nhân dẫn đến các tình trạng SMS.
Các gen thường bị xóa ở những người mắc chứng SMS đã được thu hẹp thành "vùng quan trọng" bao gồm khoảng 25 gen. Tất cả các trường hợp xóa đều bao gồm xóa gen RAI1.
Trong trường hợp không phát hiện được sự mất đoạn, cần xem xét giải trình tự của gen RAI1. Khoảng 90% các trường hợp SMS là do bị xóa, 10% còn lại là do đột biến xảy ra trong gen RAI1.
Các đột biến trong RAI1 có thể được xác định bằng trình tự sắp xếp của gen RAI1. Việc xác định trình tự RAI1 có thể được thực hiện bằng các loại xét nghiệm di truyền khác nhau. Các thử nghiệm bao gồm giải trình tự RAI1:
- Giải trình tự toàn bộ Exome: Thử nghiệm này giải trình tự tất cả các gen của một cá nhân
- Kiểm tra di truyền thông qua bảng điều khiển: Kiểm tra này chỉ bao gồm giải trình tự cho một số gen được chọn. RAI1 phải được liệt kê là một gen trong bảng. Các bảng điều trị động kinh, rối loạn phổ tự kỷ và / hoặc khuyết tật trí tuệ thường bao gồm RAI1
- Trình tự RAI1: Thử nghiệm này chỉ trình tự RAI1.

Quá trình điều trị hội chứng Smith-Magenis cần đòi hỏi có sự nỗ lực đa ngành từ các nhóm chuyên gia để xây dựng một kế hoạch điều trị thích hợp cho bệnh nhân, bao gồm bác sĩ nhi khoa, bác sĩ thần kinh, các chuyên chuyên gia phát âm và ngôn ngữ, bác sĩ tâm thần và tâm lý học.
Ngoài ra, bệnh nhân và gia đình nên tham khảo các cố vấn về di truyền trong quá trình điều trị bệnh. Hiện vẫn chưa có phương pháp điều trị rõ ràng cho những bệnh nhân mắc hội chứng Smith-Magenis ngoài trị liệu hỗ trợ và giảm nhẹ triệu chứng. Việc chẩn đoán và điều trị sớm là điều cần thiết nhằm giúp trẻ độc lập càng sớm càng tốt.
Cần giáo dục đặc biệt với liệu pháp phát âm và ngôn ngữ, vật lý trị liệu và trị liệu nghề nghiệp đối với những trẻ mắc hội chứng Smith-Magenis để giúp trẻ độc lập. Liệu pháp tích hợp cảm giác sẽ giúp trẻ đáp ứng tốt hơn với các kích thích thích do những trẻ mắc hội chứng này phản ứng rất kém.
Có thể sử dụng thuốc để điều trị các vấn đề về hành vi và tìm kiếm sự chú ý ở trẻ em mắc hội chứng Smith-Magenis. Nên tham khảo ý kiến của chuyên gia tiêu hóa kết hợp với bác sĩ nhi khoa đối với những trẻ khó khăn trong nuôi ăn. Bác sĩ có thể kê toa các thuốc chống co giật để kiểm soát các cơn co giật trong trường hợp trẻ bị động kinh.
Để đặt lịch khám tại viện, Quý khách vui lòng bấm số HOTLINE hoặc đặt lịch trực tiếp TẠI ĐÂY. Tải và đặt lịch khám tự động trên ứng dụng MyVinmec để quản lý, theo dõi lịch và đặt hẹn mọi lúc mọi nơi ngay trên ứng dụng.